 2021, Vol. 48
2021, Vol. 48文章信息
引用本文 |

作者简介
- 邓演文(1995—),男,硕士,助理工程师,研究方向为园林植物,E-mail:hinmentang@gmail.com.
通讯作者
- 曾凤(1985—),女,硕士,高级工程师,研究方向为园林植物,E-mail:pbplant@163.com.
文章历史
- 收稿日期:2021-02-22

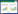


【研究意义】桃金娘科(Myrtaceae)植物主要产于亚热带和美洲的热带地区,约有100属3 000种以上,我国仅含9属约126种[1]。蒲桃属(Syzygium)为桃金娘科乔木或灌木,全球共有1 200种,我国有近80种[1]。蒲桃属植物具有一定的耐涝能力,其中蒲桃[2]、水翁[3]等均已被证实具有耐受水淹胁迫能力,可用于滨水、河畔绿化等景观用途。肖蒲桃(Syzygium acuminatissimum)为桃金娘科蒲桃属植物,原产于中国,其株型优美,嫩叶红褐色,树枝软垂,姿态优雅,兼具观果价值,适宜作为行道树或园景树[4]。目前关于肖蒲桃的研究主要在林分类型[5]、胁迫响应[6]、固氮作用[7]等方面。研究肖蒲桃的叶绿体基因组对蒲桃属的系统发育和物种鉴定具有重要意义。【前人研究进展】叶绿体参与植物的光合作用、氨基酸和脂肪酸的合成等重要生理过程,在植物生长、发育中起重要作用[8]。该细胞器具有一种环状双链DNA的遗传物质,包含100~130个基因,总长度在107~218 kb之间,并具有保守的四部分结构(一个大单拷贝区、一个小单拷贝区和两个反向重复区)[9]。由于叶绿体基因组序列高度保守、大小稳定、缺乏重组和母体遗传,因此常被用于系统发育[10]和分化时间[11]的相关研究。近年来,随着科技发展,获取基因组的成本降低,许多研究者利用叶绿体基因组数据推测植物分类学水平的系统发育关系[12]。叶绿体基因组分析技术在桃金娘科应用广泛,涵盖了桉属[13]、番樱桃属[14]、白千层属[15]、番石榴属[14]。目前,蒲桃属中,已获知海南蒲桃、丁香蒲桃和滇边蒲桃具有完整的叶绿体基因组。【本研究切入点】对于肖蒲桃的系统发育关系,有研究者仅基于3个叶绿体片段序列进行解析[16]。但简短的片段无法准确评估其在系统发育树中的位置,因此亟需通过完整的叶绿体基因组序列判定肖蒲桃在蒲桃属中的亲缘关系。【拟解决的关键问题】本研究利用高通量测序,组装和注释肖蒲桃完整的叶绿体基因组,并解析肖蒲桃叶绿体基因组结构特征与系统发育关系,旨在为蒲桃属乃至桃金娘科的系统发育研究提供依据。
1 材料与方法 1.1 试验材料肖蒲桃叶片采于广州从化百木苗场(113° 24 ′06 ″E、23 °43 ′04 ″N),植物标本(Zhang-20200729)放置于中山大学标本馆。采用CTAB法[17]对肖蒲桃叶片提取基因组DNA,-20℃下保存,备用。
1.2 试验方法1.2.1 基因组测序和注释 https://chlorobox. mpimp-golm.mpg.de/geseq.html
1.2.2 氨基酸频率、RNA编辑位点与重复序列 https://bibiserv.cebitec. uni-bielefeld.de/reputer/
1.2.3 基因组比较与序列变异分析 采用IRscope软件[27]对肖蒲桃、海南蒲桃、丁香蒲桃、滇边蒲桃4个蒲桃属叶绿体基因组中4个不同区域实现边界可视化。使用DnaSP v6.12.0软件[28]检测上述4个蒲桃属植物叶绿体基因组序列的核苷酸多样性(π)。
1.2.4 系统发育分析 为了研究肖蒲桃在桃金娘科中的系统发育关系,基于11个桃金娘科和2个菊科植物叶绿体基因组中的蛋白编码基因,使用RAxML软件[29]构建最大似然树,并设置1 000步的bootstrap。所有植物叶绿体基因组序列均从NCBI核苷酸数据库下载。
2 结果与分析 2.1 肖蒲桃叶绿体基因组结构与特征完整的肖蒲桃叶绿体基因组长度为159 352 bp,具有典型的四分结构,包括大单拷贝区(LSC) 87 993 bp,小单拷贝区(SSC)18 415 bp和一对反向重复区(IR)26 472 bp。图 1显示,肖蒲桃叶绿体基因组的总GC含量为37%,LSC、SSC和IR的GC含量分别为34.73%、30.63% 和42.66%。

|
| 图 1 肖蒲桃叶绿体基因组图谱 Fig. 1 Map of the chloroplast genome of Syzygium acuminatissimum |
本研究在肖蒲桃叶绿体基因组中共注释了109个基因,包括78个蛋白质编码基因(PCG)、27个tRNA基因和4个rRNA基因。其中,有55个基因参与自我复制,包括4个基因编码rRNA、27个基因编码tRNA、12个基因编码核糖体小亚基蛋白、8个基因编码核糖体大亚基蛋白、4个基因编码RNA聚合酶亚基;另有45个基因参与光合作用,包括6个ATP合酶基因、11个NADH脱氢酶基因、6个细胞色素b/f复合体基因、5个光系统I基因、15个光系统II基因、1个翻译起始因子IF-1和1个Rubisco长链基因(表 1)。
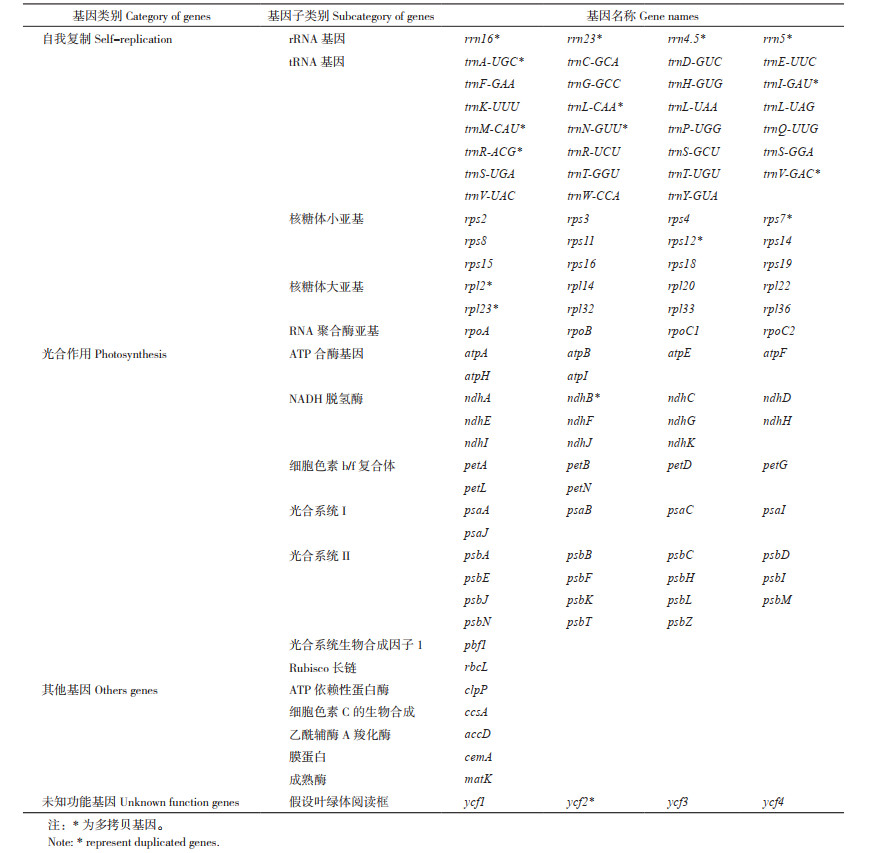
本研究在肖蒲桃叶绿体基因组中共检测到17个基因具有内含子,包括12个蛋白编码基因和5个tRNA编码基因(表 2)。ycf3、clpP、和rps12具有2个内含子,其余仅具有1个内含子。编码40S核糖体蛋白S12的rps12基因被剪接为两个片段,其中一个外显子位于大单拷贝区,另外两个外显子位于重复片段区。最长的内含子位于trnK-UUU基因(2 526 bp)中,因为其内部含有matK基因;trnL-UAA的内含子最短(530 bp)。
|
2.2 基于肖蒲桃叶绿体基因组预测的氨基酸频率、RNA编辑位点与重复序列
肖蒲桃叶绿体基因组中的蛋白编码基因共有21 379个密码子(不包含终止密码子)。由图 2可知,数量最多的3个氨基酸分别是丝氨酸(2 275)、亮氨酸(1 973)和精氨酸(1 770),而数量最少的3个分别为蛋氨酸(374)、色氨酸(485)和缬氨酸(497)。在30个最常见的密码子(RSCU>1)中,绝大多数以A或U结尾,只有UUG和AGG以G结尾。相反,在32个最不常见的密码子(RSCU<1)中,仅有UUC、CUA、AUA不以C或G结尾。此外,AUG和UGG没有密码子偏向性(RSCU=1)。

|
| 图 2 基于肖蒲桃叶绿体中78个蛋白编码基因的氨基酸频率 Fig. 2 Amino acid frequency based on 78 protein-coding genes of Syzygium acuminatissimum chloroplast |
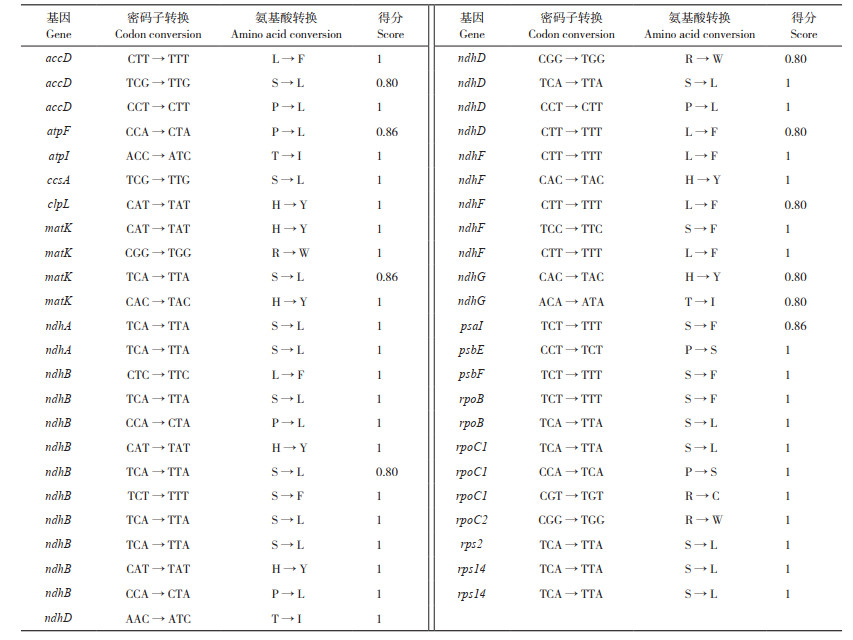
肖蒲桃叶绿体基因组中共有47个RNA可编辑位点(表 3),其中约1/3(15个)的RNA可编辑位点可将丝氨酸转化为亮氨酸。在ndhB基因检测到的RNA可编辑位点最多(10个),其次是ndhD(5个)和matK(4个)。大多数氨基酸的转化是从极性基团变为非极性基团,而只有两个位点的氨基酸基团从非极性变为极性(脯氨酸转化为丝氨酸),其中一个位于psbE基因、另一个位于rpoC1基因。
|
在肖蒲桃叶绿体基因组中共检测到48个长片段重复,其中18个正向重复、6个反向重复、22个回文重复和2个互补重复。长片段重复的长度范围在19~42 bp之间,其中长度为19 bp的重复最多(14个)、其次是22 bp(8个),而42 bp的最少(1个)。
在肖蒲桃叶绿体基因组中共检测到230个简单重复序列,其中绝大多数为单核苷酸重复(205个),其次为四核苷酸重复(12个)、三核苷酸重复(7个),双核苷酸重复(4个)和四核苷酸重复(2个)较少,未检测到六核苷酸重复。所有简单重复序列中,最长为17 bp,最短仅有8 bp。
2.3 蒲桃属叶绿体基因组比较与序列变异由图 3可知,4个蒲桃属植物的rps19基因均跨越LSC和IRb边界;rpl2基因完全位于IRb;丁香蒲桃的ndhF基因跨越IRb和SSC;ycf1基因均跨越SSC和IRa;丁香蒲桃和肖蒲桃的trnH基因跨越IRa和LSC,而海南蒲桃和滇边蒲桃的trnH基因则完全位于LSC中。

|
| 图 3 4种蒲桃属植物叶绿体基因组的4个连接边界 Fig. 3 Four junction boundaries of the chloroplast genomes of four Syzygium plants |
肖蒲桃叶绿体基因组的平均核苷酸多样性π值为0.00453,检测到7个π值较高的区域,包括trnH-psbA、 trnG-psaB、trnP-rpl33、rpl2-trnM、ndhF、ndhA、trnN-rrn23(图 4),其中2个位于基因区、5个位于基因间隔区。

|
| 图 4 4种蒲桃属植物叶绿体基因组的核苷酸多样性 Fig. 4 Nucleotide diversity of the chloroplast genomes of four Syzygium plants |
2.4 肖蒲桃的系统发育分析
为了解肖蒲桃在桃金娘科中的系统发育关系,从桃金娘科中选择11个物种作为主群体,从菊科中选择2个物种作为外类群。基于78个共有蛋白编码基因,采用RAxML构建了具有1 000个bootstrap的最大似然树(图 5),结果表明肖蒲桃与丁香蒲桃关系密切。

|
| 图 5 基于13种植物叶绿体基因组的最大似然树 Fig. 5 Maximum likelihood tree based on the chloroplast genomes of 13 species |
3 讨论
在高等植物叶绿体基因组中,通常具有长度为120~160 kb的序列、典型的四分结构[30]。肖蒲桃叶绿体基因组也不例外,其叶绿体基因组长度为159 352 bp,总GC含量为36.89%,大单拷贝、小单拷贝和反向重复区的GC含量分别为34.73%、30.63% 和42.66%。与大多数被子植物相似,反向重复区的高GC含量可能由于该区域的rRNA序列GC含量较高而引起[31]。
肖蒲桃叶绿体基因组的蛋白编码基因中共有21 379个密码子,在RSCU>1的密码子中,除了UUG外,其余密码子均以A或U结尾,这与罂粟[31]和紫荆泽兰[32]相同。在蛋白编码基因中共检测到47个可被编辑的RNA位点。其中大部分氨基酸可从丝氨酸转换为亮氨酸,而ndhB基因中的可编辑位点最多(10/47),在连翘(Forsythia suspensa) [33]和刺柏(Sanionia uncinata) [34]中也有相似研究结果。叶绿体简单重复序列是一种有效的分子标记,常用于群体遗传学、生物地理学和系统发育评估[35-36]等研究。在肖蒲桃叶绿体基因组中,绝大多数为单核苷酸重复(205/230),与大多数研究结果[37-39]一致。
由进化事件引起的反向重复区的变化导致边界和基因组大小发生细微变动,增加了物种的遗传多样性[40]。在本研究中,肖蒲桃、海南蒲桃、丁香蒲桃、滇边蒲桃4个蒲桃属植物的连接边界情况稍有不同,这可能与蒲桃属植物物种繁多、拥有丰富的遗传多样性有关[41]。
DNA条码广泛应用于植物鉴定研究[42]。然而在蒲桃属中,仅有少数几个区间用于物种鉴定,如matK、ndhF、rpl16、atpB-rbcL、trnL-F等[16, 43-44]。本研究通过计算π值发现,反向重复区比大单拷贝区和小单拷贝区区的保守性更高,该结果与其他被子植物一致[30, 45];此外,获得7个π值高于0.015的区域,包括trnH-psbA、trnG-psaB、trnP-rpl33、rpl2-trnM、ndhF、ndhA、trnN-rrn23,这些信息将为未来的物种鉴定提供依据。
蒲桃属物种繁多,为该属的物种鉴定和系统发育研究带来极大难度[1]。本研究构建的桃金娘科进化树结果与Biffin等[16]基于3个叶绿体片段得到的蒲桃属系统发育结果一致,肖蒲桃与丁香蒲桃的亲缘关系较近。但由于蒲桃属植物数量较多,目前仅有的数据并不能准确说明肖蒲桃在蒲桃属系统发育树中的准确位置,今后仍需获取更全面的数据进行深入研究分析。
4 结论本研究利用高通量测序,组装和注释了肖蒲桃完整的叶绿体基因组,并解析了该基因组的结构特征和系统发育关系,结果表明肖蒲桃叶绿体基因组的结构特征与其他蒲桃属植物相似,具有典型的四分结构,共检测到109个基因、21 379个密码子、47个RNA可编辑位点、48个长片段重复、230个简单重复序列。基因组比较分析表明,4个蒲桃属植物的IR边界有较小差异,核苷酸多样性高于0.015的区间有7个。系统发育关系分析表明,肖蒲桃与丁香蒲桃的亲缘关系较近。
| [1] |
张宏达, 缪汝愧, 陈介. 中国植物志[M]. 北京: 科学出版社, 1984, 53(1): 121-122. ZHANG H D, MIU R K, CHEN J. Flora of China[M]. Beijing: Science Press, 1984, 53(1): 121-122. |
| [2] |
薛立, 许祝莨, 李秋静, 侯晓丽, 郝云亭. 水淹胁迫对尖叶杜英和水蒲桃幼苗生理特征的影响[J]. 湖南林业科技, 2014, 41(1): 1-6. DOI:10.3969/j.issn.1009-5710.2014.01.001 XUE L, XU Z L, LI Q J, HOU X L, HAO Y T. Effects of submergence stress on physiological characteristics of Elaeocarpus apiculatus and Syzygium jambos seedlings[J]. Hunan Forestry Science & Technology, 2014, 41(1): 1-6. DOI:10.3969/j.issn.1009-5710.2014.01.001 |
| [3] |
应梦云, 冯志坚, 曹中元. 水翁幼苗耐涝试验[J]. 福建林业科技, 2013, 40(4): 79-81. DOI:10.3969/j.issn.1002-7351.2013.01.16 YING M Y, FENG Z J, CAO Z Y. The water resistance experiment of Cleistocalyx operculatus seedlings[J]. Journal of Fujian Forestry Science and Technology, 2013, 40(4): 79-81. DOI:10.3969/j.issn.1002-7351.2013.01.16 |
| [4] |
易绮斐. 肖蒲桃[J]. 热带亚热带植物学报, 2017, 25(5): 416. YI Q F. Syzygium acuminatissimum[J]. Journal of Tropical and Subtropical Botany, 2017, 25(5): 416. |
| [5] |
龙成, 杨小波, 龙文兴, 李东海. 铜鼓岭热带常绿季雨矮林5种蒲桃属植物的种群结构及空间格局[J]. 林业科学, 2015, 51(2): 18-27. DOI:10.11707/j.1001-7488.20150203 LONG C, YANG X B, LONG W X, LI D H. Population structure and spatial patterns of five Syzygium species in tropical evergreen monsoon elfin forest, Tongguling[J]. Scientia Silvae Sinicae, 2015, 51(2): 18-27. DOI:10.11707/j.1001-7488.20150203 |
| [6] |
李仕裕, 木楠, 袁晓初, 郭亚男, 于海玲, 张永夏, 王发国. 5种木本花卉对高低温的耐受性[J]. 福建林业科技, 2016, 43(1): 35-38. DOI:10.13428/j.cnki.fjlk.2016.01.007 LI S Y, MU N, YUAN X C, GUO Y N, YU H L, ZHANG Y X, WANG F G. Study on high and low temperature tolerance of 5 species ornamental trees[J]. Journal of Fujian Forestry Science and Technology, 2016, 43(1): 35-38. DOI:10.13428/j.cnki.fjlk.2016.01.007 |
| [7] |
赵亮, 周国逸, 张德强, 段洪浪, 刘菊秀. CO2浓度升高和氮沉降对南亚热带主要乡土树种及群落生物量的影响[J]. 应用生态学报, 2011, 22(8): 1949-1954. DOI:10.13287/j.101-9332.2011.0310 ZHAO L, ZHOU G Y, ZHANG D Q, DUAN H L, LIU J X. Effects of elevated CO2 concentration and nitrogen deposition on the biomass accumulation and allocation in south subtropical main native tree species and their mixed communities[J]. Chinese Journal of Applied Ecology, 2011, 22(8): 1949-1954. DOI:10.13287/j.101-9332.2011.0310 |
| [8] |
CHO K, YUN B, YOON Y, HONG S, MEKAPOGU M, KIM K, YANG T. Complete chloroplast genome sequence of tartary buckwheat (Fagopyrum tataricum)and comparative analysis with common buckwheat(F. esculentum[J]. PLOS ONE, 2015, 10(5): e125332. DOI:10.1371/journal.pone.0125332 |
| [9] |
DANIELL H, LIN C S, YU M, CHANG W J. Chloroplast genomes: diversity, evolution, and applications in genetic engineering[J]. Genome Biology, 2016, 17(1): 1-29. DOI:10.1186/s13059-016-1004-2 |
| [10] |
HU Y H, WOESTE K E, ZHAO P. Completion of the chloroplast genomes of five Chinese Juglans and their contribution to chloroplast phylogeny[J]. Frontiers in Plant Science, 2017, 7: 1955. DOI:10.3389/fpls.2016.01955 |
| [11] |
KRAK K, V T P, BELYAYEV A, DOUDA J, HREUSOV L, MAND K B. Allopolyploid origin of Chenopodium album s. str. (Chenopodiaceae): a molecular and cytogenetic insight[J]. PLOS ONE, 2016, 11(8): e161063. DOI:10.1371/journal.pone.0161063 |
| [12] |
CARBONELL-CABALLERO J, ALONSO R, IBA EZ V, TEROL J, TALON M, DOPAZO J. A phylogenetic analysis of 34 chloroplast genomes elucidates the relations between wild and domestic species within the genus Citrus[J]. Molecular Biology and Evolution, 2015, 32(8): 2015-2035. DOI:10.1093/molbev/msv082 |
| [13] |
BAYLY M J, RIGAULT P, SPOKEVICIUS A, LADIGES P Y, ADES P K, ANDERSON C, BOSSINGER G, MERCHANT A, UDOVICIC F, WOODROW I E. Chloroplast genome analysis of Australian eucalypts– Eucalyptus, Corymbia, Angophora, Allosyncarpia and Stockwellia (Myrtaceae)[J]. Molecular Phylogenetics and Evolution, 2013, 69(3): 704-716. DOI:10.1016/j.ympev.2013.07.006 |
| [14] |
RODRIGUES N F, BALBINOTT N, PAIM I, GUZMAN F, MARGIS R. Comparative analysis of the complete chloroplast genomes from six Neotropical species of Myrteae(Myrtaceae)[J]. Genetics and Molecular Biology, 2020, 43(2). DOI:10.1590/1678-4685-gmb-2019-0302 |
| [15] |
LIU H L, GENG M L, QIN Y F, XIAO Y F, LI M M. Characterization of the complete chloroplast genome of medicinal tea tree(Melaleuca alternifolia)[J]. Mitochondrial DNA Part B, 2019, 4(2): 3307-3308. DOI:10.1080/23802359.2019.1673246 |
| [16] |
BIFFIN E, CRAVEN L A, CRISP M D, GADEK P A. Molecular systematics of Syzygium and allied genera(Myrtaceae): evidence from the chloroplast genome[J]. Taxon, 2006, 55(1): 79-94. DOI:10.2307/25065530 |
| [17] |
DOYLE J, DOYLE J L. Genomic plant DNA preparation from fresh tissue-CTAB method[J]. Phytochem Bull, 1987, 19(11): 11-15. |
| [18] |
CHEN Y X, CHEN Y S, SHI C M, HUANG Z B, ZHANG Y, LI S K, LI Y, YE J, YU C, LI Z. SOAPnuke: a MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data[J]. Gigascience, 2018, 7(1): 120. DOI:10.1093/gigascience/gix120 |
| [19] |
BANKEVICH A, NURK S, ANTIPOV D, GUREVICH A A, DVORKIN M, KULIKOV A S, LESIN V M, NIKOLENKO S I, PHAM S, PRJIBELSKI A D. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing[J]. Journal of Computational Biology, 2012, 19(5): 455-477. DOI:10.1089/cmb.2012.0021 |
| [20] |
TILLICH M, LEHWARK P, PELLIZZER T, ULBRICHT-JONES E S, FISCHER A, BOCK R, GREINER S. GeSeq-versatile and accurate annotation of organelle genomes[J]. Nucleic Acids Research, 2017, 45(W1): W6-W11. DOI:10.1093/nar/gkx391 |
| [21] |
BURLAND T G. DNASTAR'S: Lasergene sequence analysis software[J]. Methods Molecular Biology, 2000(3): 71-91. |
| [22] |
ZHENG S Y, POCZAI P, HYVONEN J, TANG J, AMIRYOUSEFI A. Chloroplot: An online program for the versatile plotting of organelle genomes[J]. Frontiers in Genetics, 2020, 11. DOI:10.3389/fgene.2020.576124 |
| [23] |
KUMAR S, ST ECHER G, TAMUR A K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets[J]. Molecular Biology and Evolution, 2016, 33(7): 1870-1874. DOI:10.1093/molbev/msw054 |
| [24] |
MOWER J P. The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments[J]. Nucleic Acids Research, 2009, 37(S2): 253-259. DOI:10.1093/nar/gkp337 |
| [25] |
KURTZ S, CHOUDHURI J V, OHLEBUSCH E, SCHLEIERMACHER C, STOYE J, GIEGERICH R. REPuter: the manifold applications of repeat analysis on a genomic scale[J]. Nucleic Acids Research, 2001, 29(22): 4633-4642. DOI:10.1093/nar/29.22.4633 |
| [26] |
BEIER S, THIEL T, M NCH T, SCHOLZ U, MASCHER M. MISA-web: a web server for microsatellite prediction[J]. Bioinformatics, 2017, 33(16): 2583-2585. DOI:10.1093/bioinformatics/btx198 |
| [27] |
AMIRYOUSEFI A, HYV NEN J, POCZAI P. IRscope: an online program to visualize the junction sites of chloroplast genomes[J]. Bioinformatics, 2018, 34(17): 3030-3031. DOI:10.1093/bioinformatics/bty220 |
| [28] |
ROZAS J, FERRER-MATA A, S NCHEZ-DELBARRIO J C, GUIRAO-RICO S, LIBRADO P, RAMOS-ONSINS S E, S NCHEZ-GRACIA A. DnaSP 6: DNA sequence polymorphism analysis of large data sets[J]. Molecular Biology and Evolution, 2017, 34(12): 3299-3302. DOI:10.1093/molbev/msx248 |
| [29] |
STAMATAKIS A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies[J]. Bioinformatics, 2014, 30(9): 1312-1313. DOI:10.1093/bioinformatics/btu033 |
| [30] |
WICKE S, SCHNEEWEISS G M, DEPAMPHILIS C W, M LLER K F, QUANDT D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function[J]. Plant Molecular Biology, 2011, 76(3): 273-297. DOI:10.1007/s11103-011-9762-4 |
| [31] |
ZHOU J G, CUI Y X, CHEN X L, LI Y, XU Z C, DUAN B Z, LI Y H, SONG J Y, YAO H. Complete chloroplast genomes of Papaver rhoeas and Papaver orientale: molecular structures, comparative analysis, and phylogenetic analysis[J]. Molecules, 2018, 23(2): 437. DOI:10.3390/molecules23020437 |
| [32] |
NIE X J, LV S Z, ZHANG Y X, DU X H, WANG L, BIRADAR S S, TAN X F, WAN F H, SONG W N. Complete chloroplast genome sequence of a major invasive species, crofton weed(Ageratina adenophora)[J]. PLOS ONE, 2012, 7(5): e36869. DOI:10.1371/journal.pone.0036869 |
| [33] |
WANG W B, YU H, WANG J H, LEI W J, GAO J H, QIU X P, WANG J S. The complete chloroplast genome sequences of the medicinal plant Forsythia suspensa(Oleaceae)[J]. International Journal of Molecular Sciences, 2017, 18(11): 2288. DOI:10.3390/ijms18112288 |
| [34] |
PARK M, PARK H, LEE H, LEE B, LEE J. The complete plastome sequence of an Antarctic bryophyte Sanionia uncinata(Hedw.)Loeske[J]. International Journal of Molecular Sciences, 2018, 19(3): 709. DOI:10.3390/ijms19030709 |
| [35] |
ZENG J M, CHEN X J, WU X F, JIAO F C, XIAO B G, LI Y P, TONG Z J. Genetic diversity analysis of genus Nicotiana based on SSR markers in chloroplast genome and mitochondria genome[J]. Acta Tabacaria Sinica, 2016, 22(4): 89-97. |
| [36] |
MOHAMMAD-PANAH N, SHABANIAN N, KHADIVI A, RAHMANI M, EMAMI A. Genetic structure of gall oak(Quercus infectoria) characterized by nuclear and chloroplast SSR markers[J]. Tree Genetics & Genomes, 2017, 13(3): 70. DOI:10.1007/s11295-017-1146-8 |
| [37] |
DONG W P, XU C, LI W Q, XIE X M, LU Y Z, LIU Y L, JIN X B, SUO Z L. Phylogenetic resolution in Juglans based on complete chloroplast genomes and nuclear DNA sequences[J]. Frontiers in Plant Science, 2017, 8: 1148. DOI:10.3389/fpls.2017.01148 |
| [38] |
EBERT D, PEAKALL R. Chloroplast simple sequence repeats (cpSSRs): technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species[J]. Molecular Ecology Resources, 2009, 9(3): 673-690. DOI:10.1111/j.1755-0998.2008.02319.x |
| [39] |
ZHOU T, CHEN C, WEI Y, CHANG Y X, BAI G Q, LI Z H, KANWAL N, ZHAO G F. Comparative transcriptome and chloroplast genome analyses of two related Dipteronia species[J]. Frontiers in Plant Science, 2016, 7: 1512. DOI:10.3389/fpls.2016.01512 |
| [40] |
HE L, QIAN J, LI X W, SUN Z Y, XU X L, CHEN S L. Complete chloroplast genome of medicinal plant Lonicera japonica: genome rearrangement, intron gain and loss, and implications for phylogenetic studies[J]. Molecules, 2017, 22(2): 249. DOI:10.3390/molecules22020249 |
| [41] |
MONERUZZAMAN K M, ALEBIDI A I, AL-SAIF A M. Assessment of genetic diversity in three cultivars of Syzygium samarangense grown in Malaysia by using morphological and physiological parameters[J]. Research Journal of Biotechnology, 2012, 7(3): 16-22. |
| [42] |
ZHANG W, FAN X H, ZHU S F, ZHAO H, FU L Z. Species-specific identification from incomplete sampling: applying DNA barcodes to monitoring invasive Solanum plants[J]. PLOS ONE, 2013, 8(2): e55927. DOI:10.1371/journal.pone.0055927 |
| [43] |
WIDODO P, CHIKMAWATI T, KUSUMA Y W C. Placement of Syzygium boerlagei(Merr.)Govaerts(Myrtaceae)confirmed with atpB-rbcL intergenic spacer[J]. Biotropia, 2019, 26(1): 9-15. DOI:10.11598/btb.2019.26.1.000 |
| [44] |
SOGANDI S, TRIANDRIANI W, SAPUTRI D, SUHENDAR U. Antioxidant activity of endophytic bacterial extract isolated from clove leaf(Syzygium aromaticum L.)[J]. Journal of Agriculture and Applied Biology, 2020, 1(1): 9-17. DOI:10.11594/jaab.01.01.02 |
| [45] |
ZHONG Q W, YANG S P, SUN X M, WANG L H, LI Y. The complete chloroplast genome of the Jerusalem artichoke(Helianthus tuberosus L.)and an adaptive evolutionary analysis of the ycf2 gene[J]. Peer Journal, 2019, 7: e7596. DOI:10.7717/peerj.7596 |