 2022, Vol. 49
2022, Vol. 49文章信息
引用本文 |

基金项目
- 山东省科技攻关项目(2010GSF275);山东省教育厅科研项目(J08LH10)
作者简介
- 李洁(1997—),女,在读硕士生,研究方向为中药学,E-mail:953289297@qq.com.
通讯作者
- 徐凌川(1962—),男,教授,研究方向为中药质量控制、资源开发利用及药用真菌研究与应用,E-mail:xulingchuan518@sina.com.
文章历史
- 收稿日期:2022-03-01

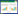


2. 山东御华景宸生态农业发展有限公司,山东 费县 273418
2. Shandong Yuhua Jingchen Ecological Agriculture Development Co. LTD, Feixian 273418, China
【研究意义】泰山白首乌(Cynanchum bungei Decne)是中药材白首乌的正品来源植物,属于萝藦科鹅绒藤属植物[1],是中国特有物种,因其善于增补人体精血,增强免疫力而延缓衰老,素有白人参的美称[2]。白首乌为多年生植物,生长周期长,野生品种的开花率和结果率均较低,人工栽培技术尚不足,种植白首乌较困难,为解决这一问题,目前关于白首乌的仿野生种植正在兴起[3]。植物的生长与土壤和环境的理化因素有关,也与其根系微环境有很大关系,尤其是根际土壤微生物,对植物的生长发育起到重要作用[4]。根际是指能受植物根系影响的范围,一般离根轴表面数毫米,是土壤- 根系- 微生物相互作用的微区域,也是不同植物种类、土壤和环境条件形成的特定的微生态系统[5]。根际微生物紧密存于根际土壤内,是根际土的重要组成部分[6],微生物群落的多样性对植物的生长有重要作用,植物生长过程中产生的分泌物也会影响微生物的种类、数量和分布,两者之间起到协同互助作用,共同促进这个微生态系统的循环[7]。【前人研究进展】研究发现,大部分根际微生物能提高土壤再利用率和促进植物生长发育,对植物可能产生的病虫害有很好的防治效果,能增强植物对外界环境的抗逆性和抗病性[8-9]。张艳琪等[10]从泰山白首乌的根际土壤中培养出具有抗菌活性的7株菌株,这也为研究开发新型抗菌药物提供了原材料的理论指导。然而,关于泰山白首乌根际土壤微生物多样性的研究,目前尚未有报道,因此,探究微生物群落的多样性,对泰山白首乌的人工栽培以及仿野生种植都有一定的理论指导意义。
【本研究切入点】随着近年来分子生物技术的发展[11],高通量测序技术被越来越多的应用于微生物多样性分析,这种方法具有准确率高、测序时间快、价格低等优势,已经成为一种新型的实验方式[12]。基于此,本研究通过Illumina MiSeq高通量测序技术,以不同产地的泰山白首乌根际土壤为研究对象,对微生物群落的组成结构和分布规律进行研究分析。【拟解决关键问题】探讨泰山白首乌的生长与微生物菌群结构变化之间的相互关系,为创造更好的仿野生种植条件、培育品质优良的泰山白首乌提供科学且有效的理论指导。
1 材料与方法 1.1 试验材料1.1.1 土壤样地条件 供试6份泰山白首乌根际土壤采自3个产地:(1)济南市莱芜区莲花山,海拔999 m,年平均气温13 ℃,年降水量1 290 mm,植被为灌木,土壤为砂壤土;(2)济南市历下区大坝头,海拔285 m,年平均气温13.8 ℃,年降水量685 mm,植被为灌木丛,土壤为砂壤土+ 岩石土;(3)济南市长清区五峰山,海拔395 m,年平均气温14.7 ℃,年降水量671 mm,植被为灌木丛,土壤为砂壤土+ 岩石土。
1.1.2 土壤样品采集 土样于2021年8月11—13日采集,采集地的泰山白首乌均为野生品种,且由山东中医药大学徐凌川教授鉴定为正品原植物。选取生长旺盛的植株,用铁铲挖至植物根部,用灭菌的刷子和不锈钢勺收集附着于根上0~4 mm范围内的土,每个样地采集地下深度10、20 cm的根际土,分别编号为LW10、LW20、LX10、LX20、CQ10、CQ20,去除大块颗粒和杂质后密封于无菌袋中,冰袋保存带回实验室,-20 ℃冰箱保存,用于后续试验。
1.2 试验方法1.2.1 根际土壤微生物DNA提取 提取DNA前,对样品进行预处理:称取300 mg混匀后的样品,放入灭菌的2 mL离心管中,加入1×PBS溶液,震荡混匀,10 000 r/min室温离心3 min,弃上层液体;将样品管放入55 ℃金属浴10 min,使残留液体完全挥发,保证后续试验操作。使用OMEGA试剂盒(E.Z.N.ATM Mag-Bind Soil DNA Kit)提取根际土壤微生物的总DNA,操作流程按照说明书进行,对提取的DNA进行琼脂糖凝胶电泳检测,并用紫外分光光度计测定其浓度和纯度,合格的DNA置于-20 ℃冰箱保存,备用。
1.2.2 PCR扩增及高通量测序 以提取的总DNA为模板,对基因组中的V3~V4区域进行扩增,通过Qubit3.0 DNA检测盒对基因组DNA进行准确定量,明确PCR反应体系中应加入的DNA量。PCR反应所采用的引物为细菌V3~V4通用引物(341F:CCTACGGGNGGCWGCAG;805R:GACTACHVGGGTATCTAATCC),反应体系按照总体积30 μL的比例配制,PCR反应程序为:94 ℃, 3 min →→(94 ℃, 30 s → 45 ℃, 20 s→ 65 ℃, 30 s)5 →→(94 ℃, 20 s → 55 ℃, 20 s→ 72 ℃, 30 s)20 →→ 72 ℃, 5 min →→ 10 ℃, ∞。PCR反应体系为:2xTrans Taq Fidelity(HiFi)PCRSuperMix 15 μL,Primer(10 μmol/L)各1 μL,Genomic DNA 1-3 μL,补充双蒸水至30 μL。PCR扩增产物经1% 的琼脂糖凝胶电泳检测,合格产物用天根胶回收试剂盒进行回收纯化,回收物送往上海生工生物股份有限公司,通过Illumina MiSeq测序平台进行高通量测序[13]。
1.2.3 生物信息学统计分析 将高通量测序结果得到的序列根据overlap关系进行拼接,区分样本后对序列质量进行质控和过滤,然后进行OTU(Operational Taxonomic Units)聚类(ASV去噪)分析和物种分类学分析[14]。基于对97% 相似水平下的OTU聚类和分类学信息,对OTU进行多样性指数分析并对群落在各个分类水平上进行统计分析,可对多个群落的结构和组成进行Alpha多样性分析、分组检验分析、差异显著性检验等一系列统计学和可视化分析[15]。最终将优化的序列根据数据库中的参考序列在门、纲、科、属的水平上进行鉴定,比较分析其群落组成、丰度以及多样性。
2 结果与分析 2.1 泰山白首乌根际土壤细菌测序分析和OTU分布对6个样品的泰山白首乌根际土壤细菌微生物进行测序分析,结果(表 1)显示,经检测合格的样品LW10、LW20、LX10、LX20、CQ10、CQ20最终有效序列片段分别为62 660、63 115、61 948、49 683、60 920、56 095条,对所有的序列以97% 的一致性进行OTUs聚类分析,共得到5 404个OTU。由软件处理得到的Veen图(图 1)可知,6个样品之间共同拥有134个OTU,不同产地地下10 cm位置的样品之间共有438个OTU,不同产地地下20 cm位置的样品之间有152个OTU,每个样品含有的OTU数不尽相同,其中样品CQ10中含有的OTU最多、为1 032个,样品LW20中含有的OTU最少、为494个。

|
| 图 1 泰山白首乌根际土壤样品间Veen图 Fig. 1 Veen diagram of rhizosphere soil samples of Cynanchum bungei Decne |
2.2 泰山白首乌根际土壤细菌丰富度和指数多样性
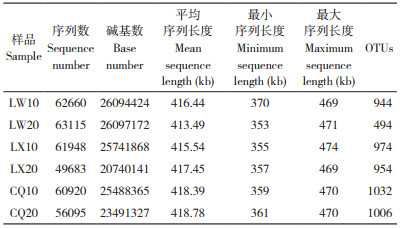
在高通量测序技术研究微生物多样性中,通过单样品的多样性分析(Alpha多样性) 可以反映微生物群落的丰度和多样性,从表 2可以看出,样品CQ10的Chao和Ace指数最大,表明该样品中微生物丰富度最高;样品LX10的Shannon指数最大且Simpson指数最小,表明该样品中微生物多样性最高;样品LX10的Shannoneven指数最大,表明该样品中微生物在群落分布的均匀度最高;6个样品的Coverage指数均为1,表明本次测序结果基本能代表泰山白首乌样品中根际细菌微生物存在的真实情况。从测序数据得到的稀释性曲线(图 2)可以看出,随着样品测序数量的增多,Alpha多样性指数值变化越来越缓慢,当达到足够的数量之后,曲线趋向于平缓,因此本次测序的深度基本能够覆盖样品根际土壤中的全部细菌微生物。Rank-abundance曲线主要是用来解释样品多样性中的多样性和均匀度,曲线越宽物种多样性越高,曲线越平坦物种组成越均匀,从测序结果(图 3)可以看出,样品LX10的多样性和均匀度略高于其他样品。
|

|
| 图 2 泰山白首乌根际土壤样品的稀释曲线 Fig. 2 Dilution curve of rhizosphere soil samples of Cynanchum bungei Decne |

|
| 图 3 泰山白首乌根际土壤样品的Rank-abundance曲线 Fig. 3 Rank-abundance curve of rhizosphere soil samples of Cynanchum bungei Decne |
2.3 泰山白首乌根际土壤细菌相关性和主成分分析
生物学重复是生物实验中常见现象,因此选择样本间相关性指标来检验测序数据的可靠性和合理性,相关系数越接近1,颜色越黄表明样本间的相似度越高,颜色越灰表示样本间相关性指数越低。由样本相关性热图(图 4)显示,相同地点不同深度的样品间相关性强,不同地点样品间相关性弱,样品LX与CQ的相关性强于样品LW与LX和CQ的相关性。由Bray-Curtis距离算法得到的样本间距离热图(图 5)可以代表样本间物种差异程度,颜色块代表距离值,颜色越灰表示物种间距离越近,可以看出相同地点不同深度的物种间距离小、不同地点物种间距离大,这与样本相关性热图相符合。

|
| 图 4 泰山白首乌根际土壤样品相关性热图 Fig. 4 Correlation heat map of rhizosphere soil samples of Cynanchum bungei Decne |

|
| 图 5 泰山白首乌根际土壤样品距离热图 Fig. 5 Distance heat map of rhizosphere soil samples of Cynanchum bungei Decne |
PCA主成分分析可以有效的找出数据中最“主要”的元素和结构,充分反映不同样本群落组成的差异和距离,不同颜色或形状的点代表不同样本,两点越接近表明两样本物种组成越相似。从图 6可以看出,不同产地对泰山白首乌根际土壤细菌微生物的多样性影响强于地下根茎深度不同的影响,验证了样本相关性和距离热图表明的不同产地间根际土壤细菌群落组成差异明显。

|
| 图 6 泰山白首乌根际土壤样品间PCA图 Fig. 6 PCA diagram of rhizosphere soil samples of Cynanchum bungei Decne |
2.4 泰山白首乌根际土壤细菌组成及分布规律
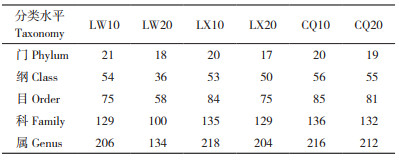
从表 3可以看出,6个样品的泰山白首乌根际土壤细菌微生物的组成和结构存在很大差异。不同产地的泰山白首乌样品根际土壤细菌分析结构显示,样品LW10、LX10、CQ10分别有21、20、20门,54、53、56纲,75、84、85目,129、135、136科,206、218、216属,表明产地因素在不同分类水平上影响泰山白首乌根际土壤微生物的多样性和丰富度;不同地下根茎深度的泰山白首乌样品根际土壤细菌分析结构显示,样品LW10、LW20分别有21、18门,54、36纲,75、58目,129、100科,206、134属,表明地下根际深度因素在不同分类水平上影响泰山白首乌根际土壤微生物的多样性和丰富度。
|
由图 7可知,3个产地6份样本中共拥17个菌门,但只有7个菌门相对丰度均大于1%,包括酸杆菌门Acidobacteria、放线菌门Actinobacteria、拟杆菌门Bacteroidetes、芽单胞菌门Gemmatimonadetes、浮霉菌门Planctomycetes、变形菌门Proteobacteria、疣微菌门Verrucomicrobia,占总土壤细菌序列的90% 以上。6份样品共拥的优势菌门为酸杆菌门和变形菌门,其中样品LW20的优势菌门为酸杆菌门,相对丰度为24.76%;样品LW10、LX10、LX20、CQ10、CQ20的优势菌门均为变形菌门,相对丰度分别为57.89%、46.00%、38.22%、27.29%、31.05%。另外,样品中均有未知菌门占一定的比例。优势物种丰度热图(图 8)也可以通过颜色直观地表达出物种的丰富度,颜色越红,丰富度越大,结果与相对丰度图符合,表明测序结果真实可靠。

|
| 图 7 泰山白首乌根际土壤细菌种类相对丰度 Fig. 7 Relative abundance of bacterial species in rhizosphere soil of Cynanchum bungei Decne |

|
| 图 8 泰山白首乌根际土壤细菌丰度热图 Fig. 8 Heatmap of bacterial abundance in rhizosphere soil of Cynanchum bungei Decne |
通过比较不同分类水平上根际土壤细菌组成(图 9)发现,在纲分类水平上,优势菌纲为放线菌纲Actinobacteria、变形菌纲Alphaproteobacteria、低温丙型变性纲Gammaproteobacteria、芽单胞菌纲Gemmatimonadetes、斯巴杆菌纲Spartobacteria、Acidobacteria-Gp4、Betaproteobacteria,其中样品LW10、LX10、LX20、CQ10的优势纲均为变形菌纲,相对丰度分别为28.48%、33.11%、24.26%、19.07;样品LW20的优势纲为斯巴杆菌纲,相对丰度为21.38%;样品CQ20的优势纲为Acidobacteria-Gp4,相对丰度为13.54%。在目分类水平上,优势菌目为微酸菌目Acidimicrobiales、放线菌目Actinomycetales、假单胞菌目Pseudomonadales、根瘤菌目Rhizobiales、鞘脂单胞菌目Sphingomonadales、Acidobacteria-Gp4、norank-Spartobacteria,其中样品LW10的优势目为假单胞菌目,相对丰度为22.39%;样品LW20的优势目为norank-Spartobacteria,相对丰度为21.38%;样品LX10的优势目为放线菌目,相对丰度为12.72%;样品LX20、CQ10的优势目均为鞘脂单胞菌目,相对丰度分别为8.02% 和10.23%;样品CQ20的优势目为Acidobacteria-Gp4,相对丰度为13.54%。在科分类水平上,优势菌科为芽单胞菌科Gemmatimonadaceae、假单胞菌科Pseudomonadaceae、红螺菌科Rhodospirillaceae、土壤红杆菌科Solirubrobacteraceae、鞘脂单胞菌科Sphingomonadaceae、Acidobacteria-Gp4、Spartobacteria,其中样品LW10的优势科为假单胞菌科,相对丰度为22.35%;样品LW20的优势科为Spartobacteria,相对丰度为21.38%;样品LX10的优势科为土壤红杆菌科,相对丰度为5.57%;样品LX20、CQ10的优势科为鞘脂单胞菌科,相对丰度分别为7.62% 和9.78%;样品CQ20的优势科为Acidobacteria-Gp4,相对丰度为13.54%。在属分类水平上,优势菌属为芽孢单菌属Gemmatimonas、Gp4、假单胞菌属Pseudomonas、土壤红杆菌属Solirubrobacter、Spartobacteria-genera-incertae-sedis、鞘氨醇单胞菌属Sphingomonas、Streptophyta,其中样品LW10的优势属为假单胞菌属,相对丰度为22.35%;样品LW20的优势属为Spartobacteria-genera-incertae-sedis,相对丰度为21.37%;样品LX10的优势属为土壤红杆菌属,相对丰度为5.57%;样品LX20的优势属为鞘氨醇单胞菌属,相对丰度为4.31%;样品CQ10的优势属为Gp4,相对丰度为9.58%;样品CQ20的优势属为Streptophyta,相对丰度为9.02%。

|
| A:纲水平;B:目水平;C:科水平;D:属水平 A: Class level; B: Order level; C: Family level; D: Genus level 图 9 泰山白首乌根际土壤细菌群落组成相对丰度 Fig. 9 Relative abundance of bacterial community composition in rhizosphere soil of Cynanchum bungei Decne |
3 讨论
土壤微生物多样性、丰富度和均匀度等指标能够影响土壤质量和植物生长状况,高通量技术作为新型测序方式,能克服传统测序技术的限制性,得到准确、系统、全面的土壤微生物群落信息[16]。有关研究报道,土壤根际微生物群落的组成和分布在一定程度上与植物种类、生长年限、海拔高度、土层深度和产地等因素有关[17]。陈冉等[18]研究发现,根际微生物群落多样性与产地因素关系密切,主要受生态区的气候和地理位置影响。官鑫等[19]研究发现,不同土层深度与土壤微生物群落的结构多样性有密切联系。本研究首次选择不同产地、不同根茎深度的泰山白首乌根际土壤样品,通过高通量16S基因Illumina MiSeq技术对土壤微生物细菌群落的组成和分布规律进行比较分析。通过对V3~V4区域测序后,共获得354 421条有效序列,平均每个样本59 071条序列,然后以97% 的相似度聚类成5 404个OTUs。通过与数据库的比较,发现不同根茎深度的样品微生物群落的组成在门水平上相似度较高,但地下10 cm位置高于地下20 cm位置处。泰山白首乌在生长成熟过程中,地下根茎会有两部分横向生长成膨大的团块状,这是其作为中药材具有最大药用价值的组织部位[20],深度约为地下10 cm和20 cm右。土壤微生物优势菌群在不同地下深度具有一定的相似性,这可能与相同植株在生长过程中适应微生物协同互助的关联性有关[21],也有研究发现这类细菌微生物具有分布广泛、适应环境强的特点,易于直接或间接地影响药用植物的生长发育和品质形成[22]。同时,由于不同土层深度的土壤碳源和有机质等理化因子存在一定差异,微生物对不同碳源的利用能力存在差异[23],因此使得不同深度的土壤微生物群落多样性存在一定的差异。
泰山白首乌产地的不同,导致其根际土壤样品中各水平微生物相对丰度有所不同。不同地域的细菌优势群落存在差异性,这可能与植株的产地生境环境有关[24],土壤理化性质的改变和生态因子的变化会导致部分微生物的生长繁殖受到限制,影响群落的丰富度、多样性和均匀度,同时微生物菌种适应环境变化的能力强弱也会影响群落多样性[25]。在各类数据比对下发现产地因素对微生物群落多样性差异的影响强于地下根茎深度对其差异的影响。
综合两个变量研究泰山白首乌根际土壤,发现变形菌门、酸杆菌门和放线菌门是各种条件下均存在的优势菌门。变形菌门是所有样品中丰富度最高的优势菌,可能是因为它们生长快、分布范围广且活动自由,易于在各类植物生长环境中存在,且该门类有多种可以进行固氮的细菌,这为植物的生长提供了有利条件[26]。酸杆菌门作为一类新型菌门,在生态系统中具有重要作用,尤其是土壤中[27]。放线菌门作为一类原核生物,大部分是腐生菌,普遍存在于土壤中,主要促进土壤中动植物遗体的腐烂和分解,并促进氮元素在自然界中的循环[28]。
植物与根际微生物之间存在着复杂的相互作用关系,根际具有极其丰富的微生物多样性,这些微生物能够影响植物的光合作用、呼吸代谢等生理过程,进而对药用植物的生长发育及根系功能产生影响,还可以通过改变药用植物根系分泌物的组成,改善土壤理化性质甚至缓解药用植物连作障碍。因此研究不同因素下根际微生物与植物的生存关系,充分利用微生物的有效促生机制以及对植物生长过程的生物防治作用,对提高植物的产量和品质具有重要的意义。充分利用新兴科技手段[29],加强对药用植物根际微生物多样性的研究,深入挖掘根际土壤菌群与植物生长的关联性,进一步探究根际微生物与药材药效成分含量的相关性,为更好地创造仿野生种植条件、培育品质优良的泰山白首乌提供科学且有效的理论指导。
4 结论本研究结果表明,6个泰山白首乌根际土壤样本共获得354 421条有效序列,平均每个样本59 071条序列,以97% 的相似度聚类成5 404个OTUs,分属于22门63纲98目170科274属。通过样品间比较,发现不同根茎深度的样品微生物群落的组成在门水平上相似度较高,但地下10 cm位置高于地下20 cm位置处,这可能是因为不同土层深度的土壤碳源和有机质等理化因子存在一定差异,深度越深,土壤成分越单一,供微生物生存的有利条件减少,且微生物对不同碳源的利用能力存在差异,造成群落多样性和丰富度的差异。3个产地中,济南市长清区的样本菌群多样性最高,莱芜区莲花山的样本菌群多样性最低,这可能与当地的土壤和气候条件有关,长清区的年降水量低,且土质类型为砂壤土+ 岩石土,适合泰山白首乌耐寒耐碱的生活条件,因此植株生长发育状况良好可以为微生物的生存提供充足且适宜的营养条件。而莱芜区的年降水量多且土壤较湿润,在一定程度上限制了泰山白首乌的生长,从而降低了根际微生物的存活。变形菌门、酸杆菌门和放线菌门是各种条件下均存在的优势菌门,这类菌群对研发泰山白首乌生长所需的生物菌肥有着重要作用。
| [1] |
陈东伟, 胡明哲, 李克明, 魏永利. 泰山白首乌的药理作用和临床应用[J]. 临床医药文献电子杂志, 2017, 4(54): 10666-10667. DOI:10.16281/j.cnki.jocml.2017.54.136 CHEN D W, HU M Z, LI K M, WEI Y L. Pharmacological action and clinical application of Cynanchum bungei Decne from Mount Tai[J]. Electronic Journal of Clinical Medical Literature, 2017, 4(54): 10666-10667. DOI:10.16281/j.cnki.jocml.2017.54.136 |
| [2] |
庄子锐, 王明亮, 彭蕴茹, 沈明勤. 白首乌C21甾苷通过TLR4通路防治大鼠肝肾纤维化的作用研究[J]. 中国中药杂志, 2021, 46(11): 2857-2864. DOI:10.19540/j.cnki.cjcmm.20200105.401 ZHUANG Z R, GU X B, PENG Y R, SHEN M Q. Effect of C21 steroidal glycoside of Cynanchum bungei Decne on prevention and treatment of liver and kidney fibrosis in rats through TLR4 pathway[J]. Chinese Journal of Traditional Chinese Medicine, 2021, 46(11): 2857-2864. DOI:10.19540/j.cnki.cjcmm.20200105.401 |
| [3] |
张明, 顾小兵, 李春阳, 邹健, 吴承东. 白首乌水旱轮作种植技术研究[J]. 农业开发与装备, 2020(11): 197-198. DOI:10.15381/j.cnki.jocml.2020.54.136 ZHANG M, GU X B, LI C Y, ZOU J, WU C D. Study on water and drought rotation planting technology of Cynanchum bungei Decne[J]. Agricultural Development & Equipments, 2020(11): 197-198. DOI:10.15381/j.cnki.jocml.2020.54.136 |
| [4] |
吴承东, 张明. 几种土壤封闭除草剂对白首乌田的除草效果试验[J]. 农业开发与装备, 2017(11): 94. WU C D, ZHANG M. Experiment on weeding effect of several soil blocking herbicides on Cynanchum bungei Decne field[J]. Agricultural Development & Equipments, 2017(11): 94. |
| [5] |
XIA A N, LIU J, KANG D C, ZHANG H G, ZHANG R H, LIU Y G. Assessment of endophytic bacterial diversity in rose by high-throughput sequencing analysis[J]. PLoS One, 2020, 15(4): e0230924. DOI:10.1371/journal.pone.0230924 |
| [6] |
WANG Z, ZHU Y, JING R, WU X Y, LI N, LIU H, ZHANG X X, WANG W P, LIU Y. High-throughput sequencing-based analysis of the composition and diversity of endophytic bacterial community in seeds of upland rice[J]. Archives of Microbiology, 2021, 203(2): 609-620. DOI:10.1007/s00203-020-02058-9 |
| [7] |
WANG Z, ZHU Y, LI N. High-throughput sequencing-based analysis of the composition and diversity of endophytic bacterial community in seeds of saline-alkali tolerant rice[J]. Microbiological Research, 2021, 250: 126794. DOI:10.1016/j.micres.2021.126794 |
| [8] |
ZHANG G F, HUANG Q L, BI X Q, LIU Y L, YUAN Z S. Analysis of endophytic bacterial community diversity and metabolic correlation in Cinnamomum camphora[J]. Archives of Microbiology, 2020, 202(1): 181-189. DOI:10.1007/s00203-019-01733-w |
| [9] |
ALEJANDRE C, HAREDER J, FUCHS B M, HEINS A. High-throughput cultivation of heterotrophic bacteria during a spring phytoplankton bloom in the North Sea[J]. Systematic and Applied Microbiology, 2020, 43(2): 126066. DOI:10.1016/j.syapm.2020.126066 |
| [10] |
张艳琪, 周甜甜, 彭川岳, 林艺伟, 王倩, 任荣康, 肖娜, 潘国军. 泰山白首乌根际土壤真菌分离纯化及抗菌活性研究[J]. 中国抗生素杂志, 2021, 46(6): 552-556. DOI:10.13461/j.cnki.cja.007074 ZHANG Y Q, ZHOU T T, PENG C Y, LIN Y W, WANG Q, REN K R, XIAO N, PAN G J. Isolation, purification and antibacterial activity of rhizosphere soil fungi from Polygonum Multiflorum Thunb[J]. Chinese Journal of Antibiotics, 2021, 46(6): 552-556. DOI:10.13461/j.cnki.cja.007074 |
| [11] |
ZHANG X, ZHANG R, GAO J, ZHENG A F. Thirty-one years of rice-rice-green manure rota-tions shape the rhizosphere microbial community and enrich beneficial bacteria[J]. Soil Biochemistry and Biote chnology, 2017, 104: 208-217. |
| [12] |
杨光柱, 黄文静, 郑丽萍, 何英云, 张宴, 孔宝华, 李云国, 赵升文, 王顺富, 李坤明, 赵丛艳, 李帆, 马钧. 基于高通量测序的苹果根腐病病株和健株根际土壤细菌组成与多样性分析[J]. 西南农业学报, 2021, 34(9): 1865-1869. DOI:10.16213/j.cnki.scjas.2021.9.008 YANG G Z, HUANG W J, ZHENG L P, HE Y Y, ZHANG Y, KONG B H, LI Y G, ZHAO S W, WANG S F, LI K M, ZHAO C Y, LI F, MA J. Analysis of bacterial composition and diversity in rhizosphere soil of apple root rot diseased and healthy plants based on high-throughput sequencing[J]. Southwest Agricultural Journal, 2021, 34(9): 1865-1869. DOI:10.16213/j.cnki.scjas.2021.9.008 |
| [13] |
陈文华. 白首乌内生真菌多样性及生物活性研究[D]. 济南: 山东中医药大学, 2020. DOI: 10.27282/d.cnki.gsdzu.2020.000521. CHEN W H. Study on diversity and bioactivity of Endophytic Fungi from Polygonum multif lorum[D]. Jinan: Shandong University of Traditional Chinese Medicine, 2020. DOI: 10.27282/d.cnki.gsdzu.2020.000521. |
| [14] |
CAPORASO J G, BITTINGER K, BUSHMAN F D. PyNAST: a flexible toolfor aligning sequences to a template alignment[J]. Bioinformatics (Oxford, England), 2010, 26(2): 266-267. DOI:10.1093/bioinformatics/btp636 |
| [15] |
WANG Q, GARRITY G M, TIEDJE J M. Naive Bayesian classifier for rapid assignment of rRNAsequences into the new bacterial taxonomy[J]. Applied and Environmental Microbiology, 2007, 73(16): 5261-5267. DOI:10.1128/AEM.00062-07 |
| [16] |
QU I JA DA N M, H EM A N DE Z M, ROR DR IGU E Z D. H ig h-throughput sequencing and food microbiology[J]. Advances in Food and Nutrition Research, 2020, 91: 275-300. DOI:10.1016/bs.afnr.2019.10.003 |
| [17] |
VERVOORT Y, LINARES A G, RONCORONI M, LIU C G, STEENSELS J, VERSTREPEN K J. High-throughput system-wide engineering and screening for microbial biotechnology[J]. Current Opinion in Biotechnology, 2017, 46: 120-125. DOI:10.1016/j.copbio.2017.02.011 |
| [18] |
陈冉, 刘志强, 王丹丹. 基于高通量测序比较不同产地药用银杏根际土壤微生物多样性[J]. 药物生物技术, 2021, 28(2): 117-122. DOI:10.27047/d.cnki.ggudu.2020.000303 CHEN R, LIU Z Q, WANG D D. Comparison of microbial diversity in rhizosphere soil of medicinal Ginkgo biloba from different habitats based on high-throughput sequencing[J]. Pharmaceutical Biotechnology, 2021, 28(2): 117-122. DOI:10.27047/d.cnki.ggudu.2020.000303 |
| [19] |
官鑫, 向阳, 祝友朋, 韩长志. 核桃根际土壤微生物群落功能多样性分析//中国植物病理学会2019年学术年会论文集[C]. 北京: 中国植物病理学会, 2019. DOI: 10.26914/c.cnkihy.2019.020569. GUAN X, XIANG Y, ZHU Y P, HAN C Z. Functional diversity analysis of soil microbial community in walnut rhizosphere//Proceedings of 2019 Annual Conference of Chinese Society of Plant Pathology[C]. Beijing: Chinese Society of Plant Pathology, 2019. DOI: 10.26914/c.cnkihy.2019.020569. |
| [20] |
印鑫, 丁永芳, 邵久针, 王明亮, 张婷, 庄子锐, 彭蕴茹. 白首乌的研究进展[J]. 中草药, 2019, 50(4): 992-1000. DOI:10.27047/d.cnki.ggudu.2019.000303 YIN X, DING Y F, SHAO J Z, WANG M L, ZHANG T, ZHUANG Z R, PENG Y R. Research progress of Polygonum multiflorum Thunb[J]. Chinese Herbal Medicine, 2019, 50(4): 992-1000. DOI:10.27047/d.cnki.ggudu.2019.000303 |
| [21] |
KHALIL M M R, FIERRO R A, PENUELAS O. Rhizospheric bacteria as potential biocontrol agents against Fusarium wilt and crown and root rot diseases in tomato[J]. Saudi Journal of Biological Sciences, 2021, 28(12): 7460-7471. DOI:10.1016/j.sjbs.2021.08.043 |
| [22] |
LI F, ZHANG X, GONG J. Specialized core bacteria associate with plants adapted to adverse environment with high calcium contents[J]. PLoS One, 2018, 13(3): e0194080. DOI:10.1371/journal.pone.0194080 |
| [23] |
SHEN C C, SHI Y, NI Y Y. Dramatic increases of soil microbial functional gene diversity at the treeline ecotone of changbai mountain[J]. Frontiers in Microbiology, 2016, 7: 1184. DOI:10.1251/frontmicrobiol.0191184 |
| [24] |
ZHANG Y G, LIU X, CONG J. The microbially mediated soil organic carbon loss under degenerative succession in an alpinemeadow[J]. Molecular Ecology, 2017, 26(14): 3676-3686. DOI:10.1418/molecularecology.0194080 |
| [25] |
GUPTA R, ANAND G, GAUR R. Plant-microbiome interactions for sustainable agriculture: a review[J]. Physiology and Molecular Biology of Plants, 2021, 27(1): 165-179. DOI:10.1007/s12298-021-00927-1 |
| [26] |
PEDERSEM B H, GURDO N, JOHANSEN HK. High-throughput dilution-based growth method enables time -resolved exo-metabolomics of Pseudomonas putida and Pseudomonas aeruginosa[J]. Microbial Biotechnology, 2021, 14(5): 2214-2226. DOI:10.1111/1751-7915.13905 |
| [27] |
OBERPAUL M, ZUMKELLER C M, CULVER T. High-throughput cultivation for the selective isolation of Acidobacteria from termite ests[J]. Frontiers in Microbiology, 2020, 11: 597628. DOI:10.3389/fmicb.2020.597628 |
| [28] |
WHITE E L, TOWER N A, RASMUSSEN L. Mycobacterium tuberculosis High-Throughput Screening[J]. Methods in Molecular Biology (Clifton, N.J.), 2016, 1439: 181-195. DOI:10.1007/978-1-4939-3673-1-12 |
| [29] |
PICHLER M, COSKUN Ö K, ORTEGA A S, NICOLA C, WORHEIDE G, VARGAS S. A 16S rRNA gene sequencing and analysis protocol for the Illumina MiniSeq platform[J]. Microbiology Open, 2018, 7(6): e00611. DOI:10.1002/mbo3.611 |
(责任编辑 邹移光)