 2023, Vol. 50
2023, Vol. 50文章信息
引用本文 |

基金项目
- 广东省乡村振兴战略专项资金种业振兴项目(2022);国家生猪产业技术体系项目(CARS-35)
作者简介
- 卢宇金(1996—),男,在读硕士生,研究方向为动物遗传育种与繁殖,E-mail:lyjin@stu.scau.edu.cn.
通讯作者
- 谢水华(1984—),男,博士,正高级畜牧师,研究方向为动物遗传育种与繁殖,E-mail:120969681@qq.com.
文章历史
- 收稿日期:2023-05-21

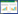


2. 华南农业大学动物科学学院,广东 广州 510642;
3. 广东艾佩克科技有限公司,广东 广州 510470
2. College of Animal Science, South China Agricultural University, Guangzhou 510642, China;
3. Guangdong iPig Technology Co., Ltd., Guangzhou 510470, China
【研究意义】Meuwissen等[1]于2001年提出了基因组选择(Genomic Selection,GS)方法,该方法使用全基因组的遗传变异及表型信息估计基因组育种值,获得了比传统育种方法更高的选择准确性。且基因组选择方法还可用于早期选种,缩短育种年限[2]。目前使用最广泛的基因组选择模型为Vanraden[3]于2008年提出基因组最佳线性无偏估计(Genomic Best Linear Unbiased Prediction, GBLUP)模型,该模型具有简单、计算效率高的特点,但在实际生产中受限于多种因素,对大量个体进行基因分型不太现实。因此,许多企业的基因分型个体与未分型个体的数量差距较大。相比于使用大量无基因型的个体作为参考群的最佳线性无偏预测(Best Linear Unbiased Prediction,BLUP)模型,利用有基因型群体的GBLUP方法未必能够获得更高的预测效果。2009年,Legarra等[4]提出了一步法基因组最佳线性无偏预测(Single-step Genomic Best Linear Unbiased Prediction, ssGBLUP)模型。该方法可同时利用系谱记录和已分型个体的基因型信息进行基因组选择,是一种能以较低成本获得更好预测效果的基因组选择方法,有利于猪、鸡等个体经济价值不高的动物进行基因组选择育种[5]。相比于GBLUP模型,ssGBLUP具有参考群规模更大的优势。而相比于传统的BLUP模型,ssGBLUP使用基因分型数据和系谱信息构建的H矩阵,更能反映个体间的加性遗传相关。因此,在合适条件下使用ssGBLUP模型进行育种,有望获得比BLUP和GBLUP更快的遗传进展。但不同群体的群体规模、遗传背景等均不同,某些条件下ssGBLUP模型未必能获得比GBLUP更高的预测准确性。且当基因分型群体规模较小时,GBLUP的预测效果并不理想,BLUP模型的预测准确性可能更高。对于群体规模较小的企业,可尝试合并相似群体的参考群进行基因组选择,而ssGBLUP模型可利用其他企业无基因型个体的数据,有望获得更高的预测准确性。【前人研究进展】除动物外,GS在作物育种中也有许多研究,如花生[6]、水稻、大麦及甘蔗等[7]。许多研究表明,GS比传统的BLUP模型的预测准确性更高。例如,Wolc等[8]对蛋鸡采食量、产蛋量的研究结果显示,基因组估计育种值的预测准确性比系谱估计育种值更高。Song等[9]对水产养殖的研究表明,GBLUP模型的预测准确性比BLUP模型更高,且加权GBLUP的预测准确性平均比GBLUP模型高6.2%。在植物疾病方面,Leplat等[10]对小麦锰缺乏症的研究也显示出GBLUP模型的预测效果高于BLUP模型。但也有研究发现,当有基因型的参考群规模较小时,GBLUP模型的预测效果并不理想[11-12]。Song等[11]对592头猪的体尺性状的研究发现,GBLUP的预测准确性低于BLUP和ssGBLUP。当有基因型个体数小于500时,ssGBLUP的预测准确性比BLUP增加的幅度较小[11]。Zhang等[12]对519只北京油鸡免疫性状的研究结果显示,该群体中GBLUP模型的预测准确性低于BLUP模型。理论上,ssGBLUP可利用无基因型个体的信息,获得比GBLUP模型更高的预测准确性。Christensen等[13]研究表明,ssGBLUP可获得比GBLUP和BLUP模型更高的准确性,Karimi等[14]以及李森[15]也在不同动物的研究中获得类似结果。但也有研究表明GBLUP的预测准确性高于ssGBLUP,如Sukhavachana等[16]对886条罗非鱼的研究结果显示,GBLUP的预测准确性高于ssGBLUP、BayesB和BayesC。
【本研究切入点】在实际育种工作中,可能由于基因型个体数较少,导致GBLUP的预测准确性不如BLUP。而ssGBLUP模型可利用无基因型个体作为参考群,提高基因组选择准确性。合并不同群体以扩大参考群规模进行联合评估,则是另一种提高基因组选择预测准确性的策略。但在合并参考群的基因组选择中,将无基因型个体纳入参考群能否提高预测准确性需要进一步探究。【拟解决的关键问题】本研究使用6家企业的大白猪群体数据,探究在不同群体及合并群体中BLUP、GBLUP及ssGBLUP模型的应用效果,并探究在合并参考群的基因组选择中,基于ssGBLUP模型将其他群体的无基因型个体纳入参考群时,对预测准确性的影响。通过对比不同群体间的遗传力、群体规模以及场间关联情况,分析不同方法在各种情形中的预测准确性。探究合并群体中无基因型个体的利用条件,为联合育种提供参考。
1 材料与方法 1.1 试验材料本研究数据来源于广东省种猪遗传评估中心,使用6家企业于2022年6月前上传的大白猪种群(分别记为A、B、C、D、E、F)数据。除C群体为法系大白外,另外5个群体为美系大白;不同群体间无精液交流,但A群体中包含7个引种于F群体的个体。研究性状为校正达100 kg体重日龄性状(DAYS_100)和校正达100 kg背膘厚性状(BFT_100)。DAYS_100测定方法参照《种猪生产性能测定规程》(NY/T 822-2019);BFT_100测定方法参照《猪活体背膘厚和眼肌面积的测定B型超声波法》(NY/T 2894-2016),使用兽用B型超声波测膘仪测定猪活体背膘。
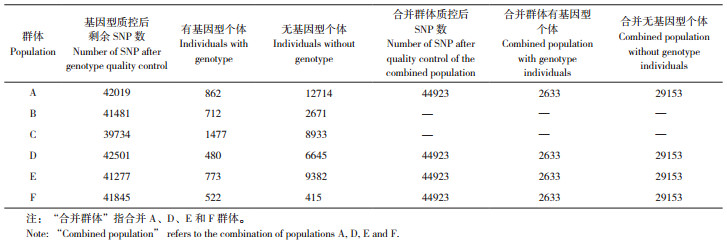
使用2款芯片进行基因分型,分别为50k中芯1号(Compass No.1 zhongxin porcine breeding chip,下称V1芯片)和57k中芯1号PLUS(KPS zhongxin-1 porcine breeding array PLUS,下称V1plus芯片)。6个研究群体的群体规模及测序情况如表 1所示。
1.2 试验方法
1.2.1 表型数据 以广东6家企业的大白猪群体的DAYS_100和BFT_100性状作为研究对象,统计各校正数据的表型均值及标准差,每个群体剔除超过群体均值±3倍标准差范围的极端数据后,用于后续分析。
1.2.2 基因型数据 本研究的部分群体使用2款芯片进行基因分型,因此需进行整理合并。首先使用PLINK软件[17]筛选常染色体位点,并提取V1和V1plus芯片的共同位点。然后,使用Beagle软件[18]分别将不同群体的基因型填充至V1plus芯片水平。基于每个群体填充后的基因型数据,使用PLINK软件进行质控。对于单群体的基因组选择,每个群体的质控标准:剔除最小等位基因频率(Minor allele frequency,maf)小于0.05的位点;剔除哈迪—温伯格平衡检验值P<10-6的位点。不同群体质控后剩余位点数在39 734~ 42 501的范围内。
使用PLINK软件进行PCA分析,并使用R软件[19]“scatterplot3d”包绘制三维PCA图像。PCA分析时,先合并6个群体填充处理后的基因型数据,并剔除重复个体。然后剔除合并群体基因型数据中maf小于0.025的位点。6个群体的合并基因型数据在质控后剩余4 822个个体及45 577个SNP。
1.3 遗传力与遗传相关使用每个群体所有个体的系谱数据和表型数据,通过DMU软件[20]的双性状BLUP模型估计每个群体的DAYS_100及BFT_100性状的遗传力及两性状间遗传相关。其中,固定效应包括出生年、出生季、出生场和性别。
1.4 场间关联率Kennedy等[21]和Mathur等[22]研究发现,最适合的场间遗传联系评价指标是不同场个体育种值的平均预测误差方差,而场效应的预测误差方差(Prediction error variance, PEV)与育种值的平均预测误差方差之间存在强相关(0.995),因此可用场效应的PEV来衡量场间关联程度的高低。根据Mathur等[22]提出的方法,场间关联率(Connectedness Rating, CR)的计算公式如下:
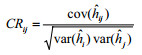
|
式中,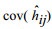
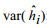

使用BLUP、GBLUP和ssGBLUP这3种模型估计育种值,对比不同模型的预测准确性。育种值的估计使用blupf 90[23]软件进行,模型如下:
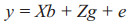
|
式中,y为性状表型值向量;b为固定效应向量(固定效应包括出生年、出生季、出生场和性别),X和Z分别为对应效应的伴随矩阵;g为随机加性遗传效应向量,即个体育种值,服从N(0, Kσg2)分布,σg2为个体加性遗传方差;e为残差向量,服从N(0, Iσe2) 分布,σe2为残差方差。其中,K为个体间亲缘关系矩阵:在BLUP模型中,矩阵K为系谱信息构建的亲缘关系矩阵A;在GBLUP中,K为基因组亲缘关系矩阵G;在ssGBLUP中,K为A矩阵与G矩阵加权构建的H矩阵。
G矩阵的构建采用Vanraden[3]提出的方法:
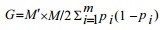
|
式中,M和M'为标准化基因型矩阵,m为SNP的数量,pi为第i个SNP的最小等位基因频率。
H矩阵根据Legarra等[4]提出的方法构建,方法如下:
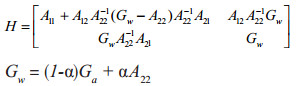
|
式中,Gw为加权构建的亲缘关系矩阵,α为系谱亲缘矩阵的权重,本研究中设α=0.01。Ga为校正后的G矩阵,根据2012年Christensen等[13]提出的方法对G矩阵进行校正。A22为已分型个体在A矩阵中的子矩阵,A11为未分型个体的亲缘关系矩阵,A12或A21为未分型个体与已分型个体间的亲缘关系矩阵。
将A、D、E和F这4个群体进行合并,探究合并其他群体参考群对预测准确性的影响。对某一群体的育种值进行预测时,不同方法所使用的群体类型如表 2所示。其中,使用合并群体有基因型个体的GBLUP方法记为C_GBLUP,使用合并群体有基因型个体及单群体无基因型个体的ssGBLUP方法记为C_ssGBLUP,使用合并群体有基因型个体及无基因型个体的ssGBLUP方法记为ssGBLUPmeg。

1.6 预测效果评估
根据使用的参考群及育种值估计模型的不同,分6种方案对比BLUP、GBLUP和ssGBLUP模型的预测准确性,每种方案均仅从单群体有基因型个体中抽取验证群,采用10次重复的十折交叉验证来评估估计育种值的预测效果。即,将每个群体的有基因型个体随机分为10份,每次选择其中1份作为验证群,其余个体作为参考群,循环10次完成一次十折交叉验证,该十折交叉验证重复10次。使用估计育种值与表型值计算预测准确性,公式如下:
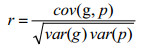
|
式中,cov(g, p)为验证群育种值与表型值的协方差,var(g)和var(p)分别为验证群育种值方差和表型值方差。
2 结果与分析 2.1 PCA分析结果6个群体4 822个基因分型个体的三维PCA分析结果(图 1)显示,6个群体聚集为3个部分,其中A、D、E和F这4个群体聚集在一起。因此,本研究选择合并这4个群体进行合并群体的基因组选择,使用PLINK软件合并这4个群体整理后的基因型数据(剔除4个重复个体后,共2 633个体),并剔除maf < 0.025的位点,质控后剩余44 923个SNP用于后续分析。

|
| 图 1 6个群体的三维PCA图 Fig. 1 Three-dimensional PCA diagram of 6 populations |
2.2 群体规模及质控后位点数统计
不同群体的群体规模及质控后的SNP位点数统计情况如表 3所示。后续研究中,评估某方案对某一群体的预测准确性时,验证群仅从该群体的有基因型个体中抽取。
2.3 遗传参数估计及场间关联率
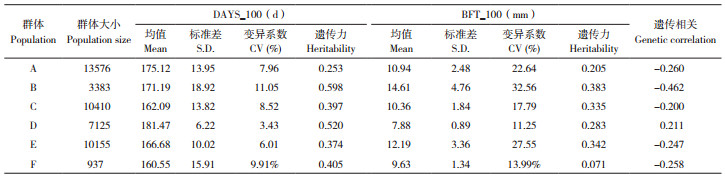
使用DMU软件,基于系谱信息估计每个群体2个生长性状的遗传力及性状间遗传相关。各群体校正后表型的描述性统计及遗传力见表 4,各群体DAYS_100性状的变异系数范围为3.43%~11.05%,BFT_100性状的变异系数范围为11.25%~32.56%。对于DAYS_100,不同群体的遗传力在0.253~0.598范围。对于BFT_100,F群体的遗传力较低,仅为0.071;其他群体BFT_100的遗传力在0.205~0.384,达中高遗传力水平。D群体的DAYS_100与BFT_100的遗传相关为0.2109,其他群体的2个生长性状间存在负的遗传相关、为-0.462~-0.247。
|
场间关联率(CR)的计算结果显示,F群体与A群体的场间关联率为3.096%,其他群体之间的场间关联率均为0。该结果与群体间的引种情况相符,A群体与F群体存在引种情况,而其他群体之间没有相互引种情况。
2.4 不同方案的预测效果评估不同方案下DAYS_100及BFT_100的预测准确性见表 5、表 6。对于DAYS_100,单群体方案预测准确性高于合并群体。其中GBLUP模型对B、C、E和F群体的预测准确性最高,分别比BLUP模型高39.22、37.09、32.44和7.78%。在有基因型个体比例低于7%的群体A和群体D中,BLUP模型的预测准确性高于GBLUP。在A、D和F群体中,ssGBLUPmeg的预测准确性低于C_ssGBLUP。
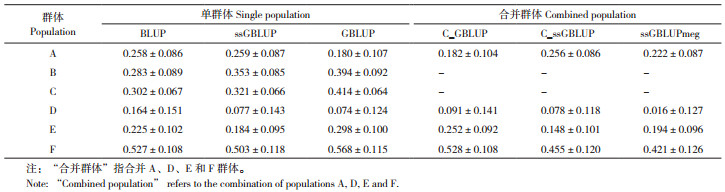
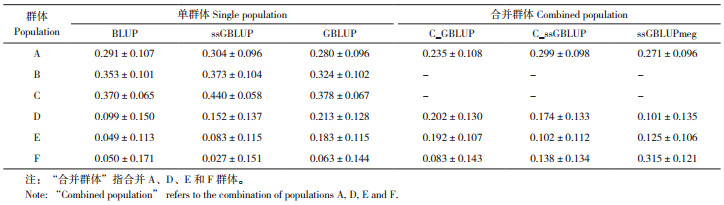
对于BFT_100性状,ssGBLUPmeg方案可以提高F群体的预测准确性,从单群体BLUP模型的0.05(±0.171)准确性提高到0.315(±0.121)。合并参考群的方案中,A群体的C_ssGBLUP方案和ssGBLUPmeg方案预测准确性高于C_GBLUP方案。但与单群体方案相比,合并参考群方案并不能提高A群体的预测准确性。对于A、B和C群体,预测准确性最高的方案为单群体ssGBLUP,分别比单群体BLUP模型提高4.47、5.67和18.92%。群体D和E中单群体GBLUP模型的预测准确性最高,分别从单群体BLUP模型的0.099(±0.15)、0.049(±0.113)提高到0.213(±0.128)和0.183(±0.115)。
3 讨论 3.1 不同群体的遗传参数达100 kg体重日龄与100 kg背膘厚性状是反映猪生长性能的重要指标。100 kg体重日龄越小说明生长速率越快,背膘厚的高低则反映瘦肉率的高低。在叶健等[24]的研究中,2个大白猪群体BFT_100性状遗传力为0.26和0.43,而DAYS_100性状遗传力为0.56和0.59。孙华等[25]的研究显示,大白猪DAYS_100性状遗传力为0.21,BFT_100性状的遗传力为0.45。贺婕妤等[26]对大白猪DAYS_100性状和BFT_100性状的遗传力研究结果为0.454和0.469。虽然不同群体的生长性状遗传力各有不同,但多数也处于中高遗传力的范围。本研究中除F群体BFT_100性状遗传力较低外,其他群体中2个生长性状的遗传力均达到中高水平。
性状间的遗传相关可以反映出2个性状同时选育的难易程度,对于存在负遗传相关的2个性状,对其中1个性状进行选育时会对另1个性状产生相反的影响。本研究发现,除D群体外,其他群体的DAYS_100性状与BFT_100性状存在负相关,遗传相关在-0.200 ~ -0.462。而D群体的DAYS_100性状与BFT_100性状的遗传相关为0.211。由于不同群体的遗传相关存在差异,合并不同群体后2个性状的遗传相关可能无法代表真实的遗传相关。因此,本研究仅使用单性状模型进行基因组选择。
3.2 不同模型的预测准确性增加有基因型个体数可提高预测准确性,进而加快遗传进展。本研究发现,在基因分型个体数>500的群体中,GBLUP或ssGBLUP模型具有较好的预测效果。对于DAYS_100性状,B、C、E和F群体预测准确性最高的模型为GBLUP,而这4个群体基因分型个体数>500且在群体中占比>7%。在有基因型个体占比<7%的A和D群体中,GBLUP模型的预测准确性低于BLUP和ssGBLUP模型。虽然群体E与群体A具有相似的群体规模,但群体A预测准确性最高的模型为ssGBLUP,而群体E中预测准确性最高的模型为GBLUP。对于BFT_100性状,A、B和C群体中ssGBLUP模型的预测准确性最高,在D、E和F群体中GBLUP模型的预测准确性最高。
ssGBLUP模型能够利用没有基因型的历史群体参与基因组选择,理论上能够获得比GBLUP模型更高的预测准确性。许多研究也显示ssGBLUP模型的预测效果优于GBLUP模型[27-32]。但也有研究显示,某些条件下ssGBLUP模型的预测效果不如GBLUP模型,例如Mrode等[33]在对奶牛体重的预测中显示GBLUP的预测准确性高于ssGBLUP模型。而周隽等[34]的研究结果显示,当有基因型个体的占比达40%~60%时,ssGBLUP模型的预测准确性高于BLUP模型,而有基因型个体占比过低时,部分性状会出现ssGBLUP模型的预测准确性低于BLUP模型的情况。这或许说明,ssGBLUP模型需要在参考群中基因分型个体数足够多且有基因型个体占比足够高的情况下,才能表现出比BLUP模型更高的预测准确性。
在本研究中,除F群体有基因型个体占比达到55.71%外,其他群体有基因型个体的占比在6.35%~21.05%。这意味着本研究中ssGBLUP模型的预测效果可能受到有基因型个体占比较低的影响,导致部分群体中ssGBLUP模型的预测准确性不如BLUP或GBLUP模型。因此,对更多个体进行测序、提高有基因型个体数有助于提高预测准确性。本研究中除C群体的有基因型个体数达到1 477外,其他群体有基因型个体数在480 ~ 862之间。由于本研究中GBLUP模型参考群规模较小,而BLUP模型参考群规模较大,部分群体会出现BLUP模型准确性更高的情况。例如,在A、D群体的DAYS_100性状中,以及A、B的BFT_100性状中,GBLUP模型的预测准确性低于BLUP模型。其他研究也表明在有基因型个体数较少,而无基因型个体数较多的情况下,BLUP模型的预测准确性可能高于GBLUP模型[11-12]。此时,使用ssGBLUP模型可能获得比BLUP模型更高的预测准确性。例如在群体A、B和C的BFT_100性状中,ssGBLUP模型的预测准确性高于BLUP和GBLUP模型。
3.3 合并参考群对预测准确性的影响当群体间场间关联率高时,合并这些群体的参考群可以提高预测准确性。本研究中,ssGBLUPmeg方案是在合并其他群体有基因型个体的基础上,同时合并其他群体无基因型个体进行一步法基因组选择。在ssGBLUPmeg方案中,F群体BFT_100性状的预测准确性大幅提高,从单群体BLUP方法0.05(±0.171)的准确性提高到0.315(±0.121)。其他群体ssGBLUPmeg方案的预测准确性与单群体ssGBLUP方案相比,相差不大或者明显下降。这或许与遗传力以及不同群体间的场间关联率(CR)有关。群体A与群体F之间的场间关联率达到3.096%,而其他群体之间的场间关联率为0。Mathur等[22]研究认为,平均场间关联率>3%的情况下进行场间的联合遗传评估较为理想。对A群体而言,F群体的群体规模较小,合并F群体带来的影响也较小。因此合并群体的基因组选择方案中A群体没有获得比单群体方案更高的预测准确性。
有研究表明,合并参考群进行基因组选择,可提高选择准确性。例如Gebreyesus等[35]对荷斯坦奶牛乳脂成分的研究中发现,合并LD结构相似的群体的参考群,可提高GBLUP的预测准确性,这与余健等[36]、张金鑫等[31]的研究结果一致。但也有研究表明,合并不同群体参考群后,基因组选择的预测准确性可能低于单群体[37-38]。在没有遗传交流的群体中,相比于单群体GBLUP,直接合并其他群体有基因型个体的GBLUP方法没有获得明显的准确性提高。这是因为不同群体间遗传结构存在各种差异,如遗传力、主效QTL和SNP的效应以及LD结构等,这些差异影响了合并群体育种值的估计。群体间的差异越小,合并群体越有可能获得更高的预测效果。针对不同群体间的差异,对基因组选择模型进行调整,有助于提高合并群体的预测准确性,如Ye等[39]采用单倍型方法、Gebreyesus等[35]采用基因组特征GBLUP模型和Schmid等[40]考虑不同群体单倍型的起源调整基因组选择模型等,提高了多群体基因组选择的预测准确性。
4 结论本研究对比了不同大白猪群体中BLUP、GBLUP及ssGBLUP模型的预测准确性,探究了联合评估中基因组选择应用效果。结果显示,在基因分型群体规模>500且群体中基因分型个体占比>7% 的群体中,基因组选择方法的预测准确性比传统BLUP模型更高。在基因分型个体占比小于7%的群体中,BLUP模型优于基因组选择方法。在存在一定遗传交流、场间关联率达到3%的群体中,采用ssGBLUP模型进行联合评估,可提高低遗传力性状的预测准确性。因此,不同企业间加强群体间遗传物质交流,进行联合育种,有助于提高部分性状的遗传进展。
| [1] |
MEUWISSEN T H, HAYES B J, GODDARD M E. Prediction of total genetic value using genome-wide dense marker maps[J]. Genetics, 2001, 157(4): 1819-1829. DOI:10.1093/genetics/157.4.1819 |
| [2] |
SCHAEFFER L R. Strategy for applying genome-wide selection in dairy cattle[J]. Journal of Animal Breeding and Genetics, 2006, 123(4): 218-223. DOI:10.1111/j.1439-0388.2006.00595.x |
| [3] |
VANRADEN P M. Efficient methods to compute genomic predictions[J]. Journal of Dairy Science, 2008, 91(11): 4414-4423. DOI:10.3168/jds.2007-0980 |
| [4] |
LEGARRA A, AGUILAR I, MISZTAL I. A relationship matrix including full pedigree and genomic information[J]. Journal of Dairy Science, 2009, 92(9): 4656-4663. DOI:10.3168/jds.2009-2061 |
| [5] |
潘荣杨, 张哲, 高宁, 陈赞谋, 李加琪, 张豪. 基因组选择一步法理论及应用研究进展[J]. 广东农业科学, 2016, 43(9): 124-131. DOI:10.16768/j.issn.1004-874X.2016.09.019 PAN R Y, ZHANG Z, GAO N, CHEN Z M, LI J Q, ZHANG H. Research progress in single step procedure theory and application in genomic selection[J]. Guangdong Agricultural Sciences, 2016, 43(9): 124-131. DOI:10.16768/j.issn.1004-874X.2016.09.019 |
| [6] |
陈小平, 鲁清, 洪彦彬, 李少雄, 梁炫强. 花生基因组学在遗传育种中的研究进展[J]. 广东农业科学, 2021, 48(12): 33-43. DOI:10.16768/j.issn.1004-874X.2021.12.005 CHEN X P, LU Q, HONG Y B, LI S X, LIANG X Q. Research progress in genomics and breeding of peanut[J]. Guangdong Agricultural Sciences, 2021, 48(12): 33-43. DOI:10.16768/j.issn.1004-874X.2021.12.005 |
| [7] |
陈雨, 姜淑琴, 孙炳蕊, 潘大建, 范芝兰, 陈文丰, 李晨. 基因组选择及其在作物育种中的应用[J]. 广东农业科学, 2017, 44(9): 1-7. DOI:10.16768/j.issn.1004-874X.2017.09.001 CHEN Y, JIANG S Q, SUN B R, PAN D J, FAN Z L, CHEN W F, LI C. Genomic selection and its applicationin on crops breeding[J]. Guangdong Agricultural Sciences, 2017, 44(9): 1-7. DOI:10.16768/j.issn.1004-874X.2017.09.001 |
| [8] |
WOLC A, ARANGO J, JANKOWSKI T, SETTAR P, FULTON J E, O'SULLIVAN N P, FERNANDO R, GARRICK D J, DEKKERS J C M. Pedigree and genomic analyses of feed consumption and residual feed intake in laying hens[J]. Poultry Science, 2013, 92(9): 2270-2275. DOI:10.3382/ps.2013-03085 |
| [9] |
SONG H L, HU H X. Strategies to improve the accuracy and reduce costs of genomic prediction in aquaculture species[J]. Evolutionary Applications, 2022, 15(4): 578-590. DOI:10.1111/eva.13262 |
| [10] |
LEPLAT F, JENSEN J, MADSEN P. Genomic prediction of manganese efficiency in winter barley[J]. The Plant Genome, 2016, 9(2). DOI:10.3835/plantgenome2015.09.0085 |
| [11] |
SONG H L, ZHANG J X, ZHANG Q, DING X D. Using different single-step strategies to improve the efficiency of genomic prediction on body measurement traits in pig[J]. Frontiers in Genetics, 2019, 9: 730. DOI:10.3389/fgene.2018.00730 |
| [12] |
ZHANG J, WANG J, LI Q H, WANG Q, WEN J, ZHAO G P. Comparison of the efficiency of BLUP and GBLUP in genomic prediction of immune traits in chickens[J]. Animals (Basel), 2020, 10(3): 419. DOI:10.3390/ani10030419 |
| [13] |
CHRISTENSEN O F, MADSEN P, NIELSEN B, OSTERSEN T, SU G. Single-step methods for genomic evaluation in pigs[J]. Animal, 2012, 6(10): 1565-1571. DOI:10.1017/S1751731112000742 |
| [14] |
KARIMI K, SARGOLZAEI M, PLASTOW G S, WANG Z Q, MIAR Y. Opportunities for genomic selection in American mink: A simulation study[J]. Public Library of Science One, 2019, 14(3): 213873. DOI:10.1371/journal.pone.0213873 |
| [15] |
李森. 不同评估方法比较金陵黄鸡饲料利用效率性状的GS选择进展[D]. 北京: 中国农业科学院, 2021. DOI: 10.27630/d.cnki.gznky.2021.000216. LI S. Assessment of GS genetic gain of feed utilizationefficiency in Jinling yellow-feathered broilers by different methods[D]. Beijing: Chinese Academy of Agricultural Sciences, 2021. DOI: 10.27630/d.cnki.gznky.2021.000216. |
| [16] |
SUKHAVACHANA S, TONGYOO P, LUENGNARUEMITCHAI A, POOMPUANG S. Optimizing genomic prediction using low-density marker panels for streptococcosis resistance in red tilapia (Oreochromis spp.)[J]. Animal Genetics, 2021, 52(5): 667-674. DOI:10.1111/age.13114 |
| [17] |
CHANG C C, CHOW C C, TELLIER L C, VATTIKUTI S, PURCELL S M, LEE J J. Second-generation PLINK: Rising to the challenge of larger and richer datasets.[J]. GigaScience, 2015, 4(1): 42-48. DOI:10.1186/s13742-015-0047-8 |
| [18] |
BRIAN L B, SHARON R B. A unified approach to genotype imputation and haplotype-phase inference for large data sets of trios and unrelated individuals[J]. American Journal of Human Genetics, 2009, 84(2): 2-5. DOI:10.1016/j.ajhg.2009.01.005 |
| [19] |
GIORGI FM, CERAOLO C, MERCATELLI D. The R language: An engine for bioinformatics and data science[J]. Life (Basel, Switzerland), 2022, 12(5): 648. DOI:10.3390/life12050648 |
| [20] |
MADSEN P, JENSEN J. A user's guide to DMU. A package for analyzing multivariate mixed models[EB/OL]. [2023.3.16], http://dmu.agrsci.dk.
|
| [21] |
KENNEDY B W, TRUS D. Considerations on genetic connectedness between management units under an animal model[J]. Journal of Animal Science, 1993, 71(9): 2341-2352. DOI:10.2527/1993.7192341x |
| [22] |
MATHUR P K, SULLIVAN B, CHESNAIS J P. Measuring connectedness: concept and application to a large industry breeding program[C]. Ottawa, Canada: Canadian Centre, 2002.
|
| [23] |
LOURENCO D, LEGARRA A, TSURUTA S, MASUDA Y, AGUILAR I, MISZTAL I. Single-step genomic evaluations from theory to practice: Using SNP chips and sequence data in BLUPF90[J]. Genes (Basel), 2020, 11(7): 790. DOI:10.3390/genes11070790 |
| [24] |
叶健, 傅金銮, 张锁宇, 叶道武, 李庆岗, 陈景明, 金娉婷, 张士媛, 王爱国. 安徽省猪育种核心群场间联系性和遗传参数估计[J]. 中国畜牧杂志, 2015, 51(18): 62-67. YE J, FU J L, ZHANG S Y, YE D W, LI Q G, CHEN J M, JIN P T, ZHANG S Y, WANG A G. Estimation of genetic parameters and connectedness in pig breeding nucleus herds of An'hui Province[J]. Chinese Journal of Animal Science, 2015, 51(18): 62-67. |
| [25] |
孙华, 宋忠旭, 李良华, 彭先文, 郭万正, 武华玉, 梅书棋. 中国大白猪SⅡ-1系主要生长和胴体性状的遗传参数估测[J]. 湖北农业科学, 2009, 48(12): 3086-3089. DOI:10.3969/j.issn.0439-8114.2009.12.055 SUN H, SONG Z X, LI L H, PENG X W, GUO W Z, WU H Y, MEI S Q. Estimation on genetic parameters of growth and carcass performance of the Chinese Yorkshire SⅡ 1 line[J]. Hubei Agricultural Sciences, 2009, 48(12): 3086-3089. DOI:10.3969/j.issn.0439-8114.2009.12.055 |
| [26] |
贺婕妤, 王斌虎, 廖柱, 谢红涛, 易国强, 刘毓文, 敖红, 唐中林. 长白和大白猪主要生长性状的遗传参数估计[J]. 畜牧兽医学报, 2021, 52(8): 2115-2123. HE J Y, WANG B H, LIAO Z, XIE H T, YI G Q, LIU Y W, AO H, TANG Z L. Estimation of genetic parameters of main growth traits in landrace and large white pigs[J]. Acta Veterinaria et Zootechnica Sinica, 2021, 52(8): 2115-2123. |
| [27] |
AFRAZANDEH M, ABDOLAHI-ARPANAHI R, ABBASI M A, KASHAN N E J, TORSHIZI R V. Comparison of different response variables in genomic prediction using GBLUP and ssGBLUP methods in Iranian Holstein cattle[J]. Journal of Dairy Research, 2022, 1-7. DOI:10.1017/S0022029922000395 |
| [28] |
NASERKHEIL M, LEE D H, MEHRBAN H. Improving the accuracy of genomic evaluation for linear body measurement traits using single-step genomic best linear unbiased prediction in Hanwoo beef cattle[J]. BMC Genetics, 2020, 21(1): 144. DOI:10.1186/s12863-020-00928-1 |
| [29] |
CESARANI A, GASPA G, CORREDDU F, CELLESI M, DIMAURO C. Genomic selection of milk fatty acid composition in Sarda dairy sheep: Effect of different phenotypes and relationship matrices on heritability and breeding value accuracy[J]. Journal of Dairy Science, 2019, 102(4): 3189-3203. DOI:10.3168/jds.2018-15333 |
| [30] |
HERRERA J R V, FLORES E B, DUIJVESTEIJN N, MOGHADDAR N. Accuracy of genomic prediction for milk production traits in Philippine dairy buffaloes[J]. Frontiers in Genetics, 2021, 12: 682576. DOI:10.3389/fgene.2021.682576 |
| [31] |
张金鑫, 唐韶青, 宋海亮, 高虹, 蒋尧, 江一凡, 弥世荣, 孟庆利, 于凡, 肖炜, 云鹏, 张勤, 丁向东. 北京地区大白猪基因组联合育种研究[J]. 中国农业科学, 2019, 52(12): 2161-2170. DOI:10.3864/j.issn.0578-1752.2019.12.013 ZHANG J X, TANG S Q, SONG H L, GAO H, JIANG Y, JIANG Y F, MI S R, MENG Q L, YU F, XIAO W, YUN P, ZHANG Q, DING X D. Joint genomic selection of Yorkshire in Beijing[J]. Scientia Acricultura Sinica, 2019, 52(12): 2161-2170. DOI:10.3864/j.issn.0578-1752.2019.12.013 |
| [32] |
唐振双, 殷东, 尹立林, 马云龙, 项韬, 朱猛进, 余梅, 刘小磊, 李新云, 邱小田, 赵书红. 猪基因组选择"两步走"策略的计算机模拟评估[J]. 中国农业科学, 2021, 54(21): 4677-4686. DOI:10.3864/j.issn.0578-1752.2021.21.016 TANG Z S, YIN D, YIN L L, MA Y L, XIANG T, ZHU M J, YU M, LIU X L, LI X Y, QIU X T, ZHAO S H. To evaluate the "two-step" genomic selection strategy in pig by simulation[J]. Scientia Acricultura Sinica, 2021, 54(21): 4677-4686. DOI:10.3864/j.issn.0578-1752.2021.21.016 |
| [33] |
MRODE R, OJANGO J, EKINE-DZIVENU C, ALILOO H, GIBSON J, OKEYO M A. Genomic prediction of crossbred dairy cattle in Tanzania: A route to productivity gains in smallholder dairy systems[J]. Journal of Dairy Science, 2021, 104(11): 11779-11789. DOI:10.3168/jds.2020-20052 |
| [34] |
周隽, 林清, 邵宝全, 任端阳, 李加琪, 张哲, 张豪. 猪群体一步法基因组选择应用效果评估[J]. 中国农业科学, 2022, 55(15): 3042-3049. DOI:10.3864/j.issn.0578-1752.2022.15.014 ZHOU J, LIN Q, SHAO B Q, REN D Y, LI J Q, ZHANG Z, ZHANG H. Evaluating the application effect of single-step genomic selection in pig populations[J]. Scientia Acricultura Sinica, 2022, 55(15): 3042-3049. DOI:10.3864/j.issn.0578-1752.2022.15.014 |
| [35] |
GEBREYESUS G, BOVENHUIS H, LUND M S, POULSEN N A, SUN D, BUITENHUIS B. Reliability of genomic prediction for milk fatty acid composition by using a multi-population reference and incorporating GWAS results[J]. Genetics Selection Evolution, 2019, 51(1): 16. DOI:10.1186/s12711-019-0460-z |
| [36] |
余健, 杨文静, 王晔, 周磊, 王楚端, 刘剑锋. 多个场联合遗传评估提高基因组选择准确性[J]. 中国畜牧杂志, 2021, 57(S1): 25-28. DOI:10.19556/j.0258-7033.20210616-10 YU J, YANG W J, WANG Y, ZHOU L, WANG C D, LIU J F. Multi-field joint genetic evaluation improves the accuracy of genome selection[J]. Chinese Journal of Animal Science, 2021, 57(S1): 25-28. DOI:10.19556/j.0258-7033.20210616-10 |
| [37] |
梁永虎. 西门塔尔牛和雪龙黑牛的混合群体基因组选择研究[D]. 北京: 中国农业科学院, 2018. LIANG Y H. Study on the multiple population genome selection based on Simmental and Xuelong beef cattle[D]. BeiJing: Chinese Academy of Agricultural Sciences, 2018. |
| [38] |
SONG H, YE S, JIANG Y, ZHANG Z, ZHANG Q, DING X. Using imputation-based whole-genome sequencing data to improve the accuracy of genomic prediction for combined populations in pigs[J]. Genetics Selection Evolution, 2019, 51(1): 58. DOI:10.1186/s12711-019-0500-8 |
| [39] |
YE H, ZHANG Z, REN D, CAI X, ZHU Q, DING X, ZHANG H, ZHANG Z, LI J. Genomic prediction using LD-based haplotypes in combined pig populations[J]. Frontiers in Genetics, 2022, 13. DOI:10.3389/fgene.2022.843300 |
| [40] |
SCHMID M, STOCK J, BENNEWITZ J, WELLMANN R. Improving the accuracy of multi-breed prediction in admixed populations by accounting for the breed origin of haplotype segments[J]. Frontiers in Genetics, 2022, 13: 840815. DOI:10.3389/fgene.2022.840815 |
(责任编辑 陈丽娥)