 2022, Vol. 49
2022, Vol. 49文章信息
引用本文 |

基金项目
- 国家自然科学基金(31500499);河南省高校科技创新人才项目(16HASTIT019)
作者简介
- 曾倩倩(1994—),女,在读硕士生,研究方向为植物发育生物学,E-mail:2604118970@qq.com.
通讯作者
- 邱宗波(1978—),女,博士,教授,研究方向为植物分子生物学,E-mail:qiuzongbo@126.com.
文章历史
- 收稿日期:2021-12-02




【研究意义】小麦(Triticum aestivum L.)在世界范围内广泛种植,除水稻外,其种植面积和产量最高,属于我国主要的商品粮和储藏粮,对推动国家粮食安全、生态环境保护和社会经济发展有重要作用[1]。伴随全球气候变化和水资源缺乏问题突出,干旱已成为目前制约小麦生长和发育的主要非生物胁迫因素之一。干旱胁迫可使植物叶片干枯萎缩、降低相对含水量、增加活性氧自由基和丙二醛含量等,进而发生膜脂过氧化,导致植物氧化损伤甚至死亡,严重阻碍了小麦的高产稳产及品质[2-3]。因此,研究小麦响应干旱胁迫的分子机理对提高小麦耐旱能力具有重要的理论和实践意义。
【前人研究进展】干旱胁迫会导致植物生理指标产生改变,其基因表达类型及表达水平也会在转录水平上发生变化。近年来,转录组测序(RNA-Seq)已成为研究植物在某一特定环境和时间下基因表达情况的重要手段,利用该技术可以获得大量逆境胁迫下植物细胞的基因表达模式信息,能够深入研究植物在分子水平对逆境胁迫的响应[4]。利用RNA-Seq技术,已在玉米、向日葵和水稻等植物中鉴定了多种响应逆境胁迫(如低温、干旱和病菌等)的相关基因。李武等[5]利用RNA-Seq技术,对耐低温发芽差异的母本及其自交系后代进行分析,从不同玉米材料中获得410个差异表达基因(Differentially Expressed Genes, DEGs),从中进一步筛选出20个可能与低温萌发密切相关的基因,为后续玉米种质资源的研究奠定基础。田雪慧等[6]利用RNA-Seq技术在低温处理百合中发现了大量响应冷害的相关基因,为揭示兰州百合抗冻分子机制奠定基础。Liang等[7]使用聚乙二醇6000对苗期向日葵进行模拟干旱胁迫处理,之后对根系及叶片进行测序,发现9个可能的抗旱相关基因,为研究向日葵干旱响应机制提供了新资源。
【本研究切入点】RNA-Seq技术的迅猛发展和生物信息学分析方法的快速改进,使得更快速高效地从各种生物中获取大量基因信息成为可能。此外,小麦基因组图谱注释信息的不断完善,为运用转录组分析方法探索小麦抗旱分子机制提供了研究基础。【拟解决的关键问题】本研究利用高通量RNA-Seq技术,对自然干旱处理的小麦幼苗叶片进行测序分析,挖掘小麦干旱胁迫应答的关键基因,解析小麦干旱胁迫的分子机制,丰富植物抗旱基因组数据,为后续研究提供更多的遗传资源。
1 材料与方法 1.1 试验材料供试小麦品种为黄淮麦区大面积推广的郑麦366号,由河南省农业科学院提供。试验在河南师范大学生命科学学院的光照培养室进行。2021年9月,选取形态饱满、大小均匀的小麦种子,用0.1%HgCl2消毒2 min后用去离子水冲洗干净,播种在铺有2层滤纸的培养皿中,置于25 ℃恒温培养箱中催芽。待出芽后,选取长势一致的小麦幼苗分别移栽至营养土和蛭石混合的塑料花盆中(40棵/盆,10盆),培养于人工气候室中,生长温度25 ℃/18 ℃(昼/夜),相对湿度75%,光照周期12 h/12 h,光照强度400 μmol/m2·s,浇以自来水。待幼苗长至二叶一心时,5盆幼苗继续浇以自来水(对照),另5盆幼苗停止浇水(自然干旱处理)。在停止浇水后6 d(土壤相对含水量为50%),分别取对照和自然干旱处理小麦幼苗的叶片,用液氮速冻后于-80 ℃冰箱中保存。
1.2 试验方法1.2.1 丙二醛和相对含水量的测定 参照Niu等[8]的方法测定小麦幼苗叶片的丙二醛(MDA)含量,参照Wasaya等[9]的方法检测叶片相对含水量。
1.2.2 RNA提取、cDNA文库构建及测序 对照和干旱处理小麦幼苗叶片总RNA由北京诺禾致源生物公司采用TRIzol法提取并完成质量分析和cDNA文库构建,利用Illumina HiSeq 2500平台进行RNA-Seq测序。
1.2.3 数据组装及DEGs筛选 过滤原始序列(raw reads)得到高质量的干净序列(clean reads)。利用Trinity软件组装clean reads,之后使用TGICL软件聚类去冗余得到单基因簇(Unigene)。利用FPKM值代表基因表达量水平。根据|log2 (Fold change)| > 1且FDR < 0.05筛选DEGs,并进行DEGs的GO功能分类(http://www.geneontology.org/) 与KEGG代谢通路富集(http://www.kegg.jp/kegg/kaas/) 分析。
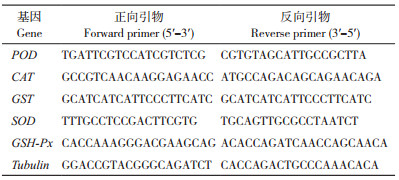
1.2.4 荧光定量PCR验证 为验证RNA-Seq测序结果的准确性,挑选5个与干旱胁迫相关的DEGs,运用Primer 5.0软件设计引物(表 1)。采用植物总RNA提取试剂盒(RNAprep Pure Plant Kit,天根生化科技有限公司)获得小麦叶片总RNA,用反转录试剂盒(Takara公司)合成cDNA。实时荧光定量PCR(quantitative real-tirne PCR, qRT-PCR)采用Rotor-Gene 3000 Real-Time PCR System(美国)进行PCR扩增,每次试验设3个生物学重复。反应体系参照SYBR® Premix Ex TaqTM Ⅱ试剂盒(Takara公司)说明书进行。以小麦Tubulin(GenBank登录号:HQ171897)为内参基因,扩增结果用2-ΔΔCt方法进行计算。
2 结果与分析 2.1 干旱处理对小麦幼苗叶片丙二醛含量和相对含水量的影响
从图 1可以看出,干旱处理后的小麦叶片MDA含量显著增加,比对照增加20.36%,相对含水量显著降低,比对照下降11.41%,说明此时小麦的细胞膜已受到严重损伤。

|
| 柱上小写英文字母不同者表示差异显著 Columns marked with lowercase letters represent significant differences 图 1 干旱处理对小麦幼苗丙二醛含量和相对含水量的影响 Fig. 1 Effects of drought stress on MDA content and relative water content in wheat seedlings |
2.2 小麦转录组测序与数据组装
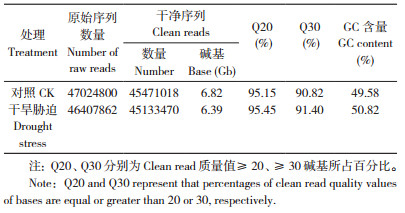
为进一步探索小麦幼苗在转录组水平上对干旱胁迫的应答,采用Illumina HiSeq 2500平台将对照和干旱处理的小麦叶片进行RNA-Seq。数据经质量控制后,对照获得45 471 018个clean reads片段,包含6.82 Gb碱基信息。干旱处理获得45 133 470个clean reads片段,包含6.39 Gb碱基信息,GC含量分别为49.58% 和50.82%,Q30(测序错误率小于0.1%)均在90% 以上(表 2)。说明Illumina平台所得到的小麦转录组序列数量和质量均较高,为后续组装和分析提供了较好的数据。
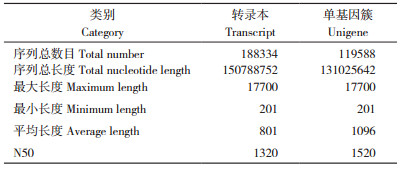
利用Trinity软件对测序所得的数据进行拼接组装,分别得到188 334个转录本和119 588个Unigene。转录本序列总长度为150 788 752 bp,平均长度为801 bp,N50为1 320 bp。Unigene总长度为131 025 642 bp,平均长度为1 096 bp,N50为1 520 bp,最长的Unigene为17 700 bp,最短的为201 bp(表 3)。Unigene的N50长度比其平均长度增加约40%,而转录本的N50长度比其平均长度增加65%,说明数据组装质量较好。
2.3 干旱处理对小麦叶片差异表达基因的影响
以FDR < 0.05且|log2 Fold change| > 1作为筛选条件,对获得的对照和干旱胁迫处理小麦叶片的DEGs分析结果用火山图展示。结果(图 2)表明,对照与干旱胁迫处理相比,共获得1 207个DEGs,其中752个上调、455个下调,上调基因在数量上高于下调基因。

|
| 红色点表示表达量上调DEGs;蓝色点表示表达量下调DEGs;灰色点表示没有差异基因 Red dots represent differential genes up-regulated in expression; blue dots represent differential genes down-regulated in expression; gray dots represent no differential genes 图 2 对照与干旱处理小麦叶片DEGs的火山图 Fig. 2 Volcano plots of DEGs in wheat leaves between control and drought treatment groups |
2.4 小麦叶片差异表达基因的GO与KEGG富集分析
本研究中1 207个DEGs获得GO功能分类,DEGs上、下调统计结果见图 3。由图 3可知,上调的DEGs中,主要归类为代谢过程和甘油代谢过程(生物过程类别)、膜和细胞部分(细胞组分)、水解酶活性和催化活性(分子功能);下调的DEGs中,主要归类为代谢过程和主要代谢过程(生物过程)、细胞器和细胞(细胞组分)、催化活性和氧化还原酶活性(分子功能)。整体上来看,GO富集分析在绝大部分同一条目下富集的DEGs中上调基因数量多于下调基因数量,但在细胞器和细胞条目中下调基因数量高于上调基因数量。

|
| A:生物过程;B:细胞组分;C:分子功能 A: Biological process; B: Cell component; C: Molecular function 图 3 小麦叶片DEGs的GO功能分类 Fig. 3 GO classification of DEGs in wheat leaves |
经KEGG富集显示,两组样品的1 207个DEGs共富集得到65条信号通路,其中有9条信号通路富集显著(P < 0.05),主要包括淀粉和蔗糖代谢(富集的DEGs数最多)、卟啉和叶绿素代谢、光合作用-天线蛋白、光合作用、苯丙烷生物合成、乙醛酸和二羧酸代谢、甘油磷脂代谢、氨基酸代谢、光合生物中的碳固定等(图 4)。

|
| 圆点所在位置表示富集的条目,圆点大小代表DEGs的数量 The position of the dot represents the enriched item, and the size of the dot represents the number of DEGs 图 4 小麦叶片DEGs的KEGG富集分析 Fig. 4 KEGG enrichment analysis of DEGs in wheat leaves |
2.5 小麦叶片差异表达基因的荧光定量验证
为了验证RNA-seq结果的准确性,选择5个抗氧化酶基因进行qRT-PCR检测。由图 5可见,5个基因的表达变化情况与RNA-Seq一致,表明测序结果较为准确、可靠。此外,5个抗氧化酶基因均上调表达,且POD和CAT基因的表达变化水平高于SOD、GST和GSH-Px,说明干旱胁迫可以诱导抗氧化酶基因及相关物质的表达,且POD和CAT基因可能起主导作用,促进小麦对活性氧的清除,降低活性氧对细胞的膜系统伤害。

|
| 图 5 干旱处理小麦叶片DEGs的qRT-PCR表达分析 Fig. 5 qRT-PCR analysis of DEGs in wheat leaves under drought stress |
3 讨论
干旱对植物的生长发育、生理代谢、生殖发育等多种生物过程均会造成不利影响,是限制粮食作物品质和产量的主要逆境灾害之一[10-11]。研究发现,干旱胁迫后,植物可在生理生化和分子水平方面作出胁迫响应以抵御逆境损伤,降低环境伤害[12-13]。植物受到干旱刺激后,能够诱导体内的渗透调节系统调控逆境胁迫应答,相对含水量在渗透调节过程中具有重要作用,可以反映植物对干旱的耐受性[14]。Meher等[14]用不同浓度的PEG6000处理花生24 h,结果表明,随着PEG6000浓度的增加,叶片和根中相对含水量均显著降低。此外,胡艳等[15]研究发现,随着土壤干旱胁迫的逐渐加剧,黑果腺肋花楸叶片相对含水量逐渐降低。本研究结果与该结果一致,间接反映了植物的气孔状况和光合功能受到影响。植物在逆境胁迫下会诱导生成活性氧,这些活性氧与生物大分子(脂质和蛋白质等)发生反应,最终生成MDA,造成膜伤害,进而影响植物代谢过程[14]。本研究中,小麦在干旱胁迫下叶片的MDA含量显著高于对照,说明小麦细胞膜损伤严重。
当植物受到胁迫时,基因的表达模式和表达水平会发生显著变化,通过挖掘和筛选调控干旱的基因,可以为研究植物适应逆境提供新的思路和方法。RNA-Seq是近年发展起来的用来鉴定差异表达基因的技术,是筛选抗逆基因的重要研究手段[16-17]。本研究采用Illumina HiseqTM 2500高通量测序平台对干旱处理和对照的小麦叶片进行了RNA-Seq,获得的平均样本数据量为6.82 Gb,通过Trinity拼接组装得到18 334个转录本和119 588个Unigene,共筛选获得1 207个DEGs,其中752个上调,455个下调,上调基因在数量上高于下调基因,可见,差异基因主要以上调表达为主。
植物响应干旱胁迫是一个受多个基因控制,涉及多种途径和多种代谢产物合成的复杂过程[18]。本研究中,GO富集发现,小麦中检测到的1 207个DEGs富集到3个GO本体的34个条目下,整体来看,主要富集在水解酶活性、催化活性、代谢过程、甘油代谢过程、膜和细胞部分等,GO分析在绝大部分同一条目下富集的DEGs中上调基因数多于下调基因,说明小麦主要通过上调基因表达来响应干旱胁迫,同时也说明干旱环境对小麦的胁迫作用是多方面、复杂的。KEGG通路富集发现,DEGs显著富集在淀粉和蔗糖代谢、卟啉和叶绿素代谢、光合作用-天线蛋白、光合作用、苯丙烷生物合成、乙醛酸和二羧酸代谢、甘油磷脂代谢、氨基酸代谢、光合生物中的碳固定等通路。在干旱条件下,植物光合作用-天线蛋白、叶绿体组分、氨基酸代谢和碳代谢等通路受到影响,使得光合作用和物质积累受到抑制,这可能是小麦叶片相对含水量降低及MDA含量增加的主要原因。
研究表明,植物在逆境胁迫下体内会产生对自身有害的活性氧,一些抗氧化酶(如SOD和CAT等)及抗氧化剂具有清除活性氧的功能,能够使其产生与清除达到平衡状态,使之抵抗逆境胁迫,维持植物的正常生长[19-20]。RNA-Seq结果显示,干旱条件下,富集在POD和CAT活性等功能组的多个DEGs表达上调,可以促进抗氧化酶及相关物质的产生,提高小麦对活性氧的清除能力,从而降低活性氧对细胞的膜系统伤害。作为一种非酶促活性氧清除剂,谷胱甘肽类似于抗氧化酶,可以清除干旱胁迫诱导的自由基和过氧化物[21]。本研究中,编码GST和GSH-Px的DEGs在干旱胁迫下表达均上调,进而调控谷胱甘肽的代谢过程,对于清除细胞代谢过程中累积的自由基,减轻细胞所受的干旱胁迫损伤具有重要作用。
4 结论本研究对正常生长与自然干旱处理下郑麦366号的叶片进行转录组分析,共获得1 207个差异表达基因,其中752个表达上调,455个表达下调。GO功能富集分析显示,DEGs主要富集在水解酶活性、催化活性、代谢过程、甘油代谢过程、膜和细胞部分、细胞和氧化还原酶活性等。KEGG途径富集分析发现,DEGs显著富集于淀粉和蔗糖代谢、卟啉和叶绿素代谢、光合作用-天线蛋白、光合作用、苯丙烷生物合成、乙醛酸和二羧酸代谢、甘油磷脂代谢、氨基酸代谢和光合生物中的碳固定等代谢通路。qRT-PCR分析结果表明,5个抗氧化酶基因的表达趋势和RNA-Seq测序结果一致。此外,还候选了POD、POD、CAT及GST基因作为小麦自然干旱胁迫响应相关的应答候选基因。
| [1] |
王禹. 新形势下我国粮食安全保障研究[D]. 北京: 中国农业科学院, 2016. WANG Y. Research on food security in China under the new situation[D]. Beijing: Chinese Academy of Agricultural Sciences, 2016. |
| [2] |
AHMAD Z, WARAICH E A, AKHTAR S, ANJUM S, AHMAD T, MAHBOOB W, HAFEEZ O B A, TAPERA T, LABUSCHAGNE M, RIZWAN M. Physiological responses of wheat to drought stress and its mitigation approaches[J]. Acta Physiologiae Plantarum, 2018, 40(4): 1-13. DOI:10.1007/s11738-018-2651-6 |
| [3] |
LANGRIDGE P, REYNOLDS M. Breeding for drought and heat tolerance in wheat[J]. Theoretical and Applied Genetics, 2021, 134(6): 1-17. DOI:10.1007/s00122-021-03795-1 |
| [4] |
CHAICHI M, SANJARIAN F, RAZAVI K, GONZALEZ-HERNANDEZ J L. Analysis of transcriptional responses in root tissue of bread wheat landrace (Triticum aestivum L.) reveals drought avoidance mechanisms under water scarcity[J]. PLOS ONE, 2019, 14(3): e0212671. DOI:10.1371/journal.pone.0212671 |
| [5] |
李武, 李光玉, 肖颖妮, 胡建广, 李春艳, 李余良, 卢文佳, 李高科, 刘建华. 基于转录组测序解析甜玉米耐低温发芽的相关基因[J]. 广东农业科学, 2021, 48(3): 11-18. DOI:10.16768/j.issn.1004-874X.2021.03.002 LI W, LI G Y, XIAO Y N, HU J G, LI C Y, LI Y L, LU W J, LI G K, LIU J H. Analysis of genes related to low-temperature germination ability in sweet corn based on transcriptome sequencing[J]. Guangdong Agricultural Sciences, 2021, 48(3): 11-18. DOI:10.16768/j.issn.1004-874X.2021.03.002 |
| [6] |
田雪慧, 郁继华, 颉建明. 转录组测序研究兰州百合抗冻关键基因及途径[J]. 广东农业科学, 2019, 46(8): 35-43. DOI:10.16768/j.issn.1004-874X.2019.08.006 TIAN X H, YU J H, XIE J M. Study on key anti-freeze genes and pathways of Lanzhou lily (Lilium davidii var. unicolor) by transcriptome sequencing[J]. Guangdong Agricultural Sciences, 2019, 46(8): 35-43. DOI:10.16768/j.issn.1004-874X.2019.08.006 |
| [7] |
LIANG C B, WANG W J, WANG J, MA J, LI C, ZHOU F, ZHANG S Q, YU Y, ZHANG L G, LI W Z, HUANG X T. Identification of differentially expressed genes in sunflower (Helianthus annuus) leaves and roots under drought stress by RNA sequencing[J]. Botanical Studies, 2017, 58(1): 1-11. DOI:10.1186/s40529-017-0197-3 |
| [8] |
NIU T, ZHANG T, QIAO Y, WEN P, ZHAI G, LIU E, AL-BAKRE D A, AL-HARBI M S, GAO X, YANG X. Glycinebetaine mitigates drought stress-induced oxidative damage in pears[J]. PLOS ONE, 2021, 16(11): e0251389. DOI:10.1371/journal.pone.0251389 |
| [9] |
WASAYA A, MANZOOR S, YASIR T A, SARWAR N, MUBEEN K, ISMAIL I A, RAZA A, REHMAN A, HOSSAIN A, EL SABAGH A. Evaluation of fourteen bread wheat (Triticum aestivum L.) genotypes by observing gas exchange parameters, relative water and chlorophyll content, and yield attributes under drought stress[J]. Sustainability, 2021, 13(9): 4799. DOI:10.3390/su13094799 |
| [10] |
LI W M, WANG Y J, ZHANG Y B, WANG R Y, GUO A H, XIE Z K. Impacts of drought stress on the morphology, physiology, and sugar content of Lanzhou lily (Lilium davidii var. unicolor)[J]. Acta Physiologiae Plantarum, 2020, 42(8): 1-11. DOI:10.1007/s11738-020-03115-y |
| [11] |
TAMILSELVI C, VENKATESHWARAN R, JEEVAPRIYA S, ANITHA R, ARULSELVI S. Physiological studies on the effect of plant growth regulators for mitigation of drought in rice (Oryza sativa L.)[J]. Journal of Pharmacognosy and Phytochemistry, 2021, 10(2): 1035-1041. |
| [12] |
RAZI K, MUNEER S. Drought stress-induced physiological mechanisms, signaling pathways and molecular response of chloroplasts in common vegetable crops[J]. Critical Reviews in Biotechnology, 2021, 41(5): 1-40. DOI:10.1080/07388551.2021.1874280 |
| [13] |
SAPES G, SALA A. Relative water content consistently predicts drought mortality risk in seedling populations with different morphology, physiology, and times to death[J]. Plant, Cell and Environment, 2021, 44(10): 3322-3335. DOI:10.1111/pce.14149 |
| [14] |
MEHER, SHIVAKRISHNA P, REDDY K A, RAO D M. Effect of PEG6000 imposed drought stress on RNA content, relative water content (RWC), and chlorophyll content in peanut leaves and roots[J]. Saudi Journal of Biological Sciences, 2018, 25(2): 285-289. DOI:10.1016/j.sjbs.2017.04.008 |
| [15] |
胡艳, 艾力江·麦麦提, 安尼瓦尔·艾木都, 古丽买然木·吐尼亚孜, 阿不都外力·吐尔洪, 买尔开那·赛买提江. 土壤逐渐干旱及复水对黑果腺肋花楸光合特性的影响[J]. 广东农业科学, 2020, 47(1): 39-47. DOI:10.16768/j.issn.1004-874X.2020.01.006 HU Y, MAIMAITI A, AIMUDU A, TUNIYAZI G, TUERHONG A, SAIMAITIJIANG M. Response of photosynthetic performance in Aronia melanocarpa to drought stress and rehydration[J]. Guangdong Agricultural Sciences, 2020, 47(1): 39-47. DOI:10.16768/j.issn.1004-874X.2020.01.006 |
| [16] |
MUTHUSAMY M, UMA S, BACKIYARANI S, SARASWATHI M S, CHANDRASEKAR A. Transcriptomic changes of drought-tolerant and sensitive banana cultivars exposed to drought stress[J]. Frontiers in Plant Science, 2016, 7: 1609. DOI:10.3389/fpls.2016.01609 |
| [17] |
SONG K, KIM H C, SHIN S, KIM K-H, MOON J-C, KIM J Y, LEE B-M. Transcriptome analysis of flowering time genes under drought stress in maize leaves[J]. Frontiers in Plant Science, 2017, 8: 267. DOI:10.3389/fpls.2017.00267 |
| [18] |
WEI B, HOU K, ZHANG H, ZHANG H H, WANG X Y, WU W. Integrating transcriptomics and metabolomics to studies key metabolism, pathways and candidate genes associated with drought-tolerance in Carthamus tinctorius L. under drought stress[J]. Industrial Crops and Products, 2020, 151: 112465. DOI:10.1016/j.indcrop.2020.112465 |
| [19] |
ZHANG C M, SHI S, LIU Z, YANG F, YIN G L. Drought tolerance in alfalfa (Medicago sativa L.) varieties is associated with enhanced antioxidative protection and declined lipid peroxidation[J]. Journal of Plant Physiology, 2019, 232: 226-240. DOI:10.1016/j.jplph.2018.10.023 |
| [20] |
ZHANASSOVA K, KURMANBAYEVA A, GADILGEREYEVA B, YERMUKHAMBETOVA R, IKSAT N, AMANBAYEVA U, BEKTUROVA A, TLEUKULOVA Z, OMAROV R, MASALIMOV Z, MASALIMOV Z. ROS status and antioxidant enzyme activities in response to combined temperature and drought stresses in barley[J]. Acta Physiologiae Plantarum, 2021, 43(8): 1-12. DOI:10.1007/s11738-021-03281-7 |
| [21] |
王玮玮, 唐亮, 周文龙, 杨燕, 高波, 赵云峰, 王伟. 谷胱甘肽生物合成及代谢相关酶的研究进展[J]. 中国生物工程杂志, 2014, 34(7): 89-95. DOI:10.13523/j.cb.20140714 WANG W W, TANG L, ZHOU W L, YANG Y, GAO B, ZHAO Y F, WANG W. Progress in the biosynthesis and metabolism of glutathione[J]. China Biotechnology, 2014, 34(7): 89-95. DOI:10.13523/j.cb.20140714 |
(责任编辑 崔建勋)