 2022, Vol. 49
2022, Vol. 49文章信息
引用本文 |

基金项目
- 广东省农业科学院青年导师制项目(R2020QD-021);广东省农业科学院院长基金(202026);广东省重点领域研发计划项目(2020B020223004)
作者简介
- 常虹(1989—),女,博士,助理研究员,研究方向为昆虫分子生物学、害虫防治等,E-mail:changyan305@163.com.
通讯作者
- 孙海滨(1962—),男,研究员,研究方向为农药应用、农药环境行为等,E-mail:sunhb@gdppri.cn.
文章历史
- 收稿日期:2021-10-15




【研究意义】黄地老虎(Agrotis segetum Denis and Schiffermüller)是一种重要的世界性农业害虫,隶属于鳞翅目(Lepidoptera)夜蛾科(Noctuidae),广泛分布于亚洲、欧洲和非洲等地。黄地老虎主要为害棉花、马铃薯、玉米和十字花科蔬菜等作物,可对我国农业和经济造成较大损失[1-2]。同时,它也是一种重要的迁飞害虫,已有研究表明其在我国进行季节性迁飞[3-4]。迁飞可促进种群间的基因交流,极大降低不同地理种群间遗传分化的程度。对黄地老虎种群遗传结构进行研究,有助于探讨不同地理种群间的遗传分化关系,揭示其迁飞路径及起源,进而为明确该虫的种群发生动态及成灾机制提供科学依据。【前人研究进展】微卫星又称简单重复序列(simple sequence repeats,SSR),是一类由1~6个核苷酸简单串联重复组合而形成的核苷酸序列,一般由中心的重复序列和两端的保守序列构成。微卫星具有共显性遗传、多态性信息含量高、具有很高的突变速率及稳定性高、实验重复性好等优点,被广泛应用于种群遗传结构及遗传分化等种群遗传学的研究[5-8]。单核苷酸多态性(single nucleotide polymorphism,SNP)主要是指在基因组水平上由单个核苷酸变异引起的DNA序列多态性,具有分布广泛、数量丰富、遗传稳定性高、较高DNA降解耐受性等特点,被用于群体遗传分析及品种鉴定等研究[9-12]。传统的SSR和SNP分子标记的开发需要构建基因组文库进行克隆测序,检测过程繁琐且费时费力。随着高通量测序技术的快速发展,转录组测序技术为非模式生物SSR和SNP位点的挖掘提供了一种高效、快速、经济的途径。因此,基于转录组数据筛选SSR和SNP位点被广泛应用于动植物的研究中[13-16]。黎东海等[13]基于齿缘刺猎蝽Sclomina erinacea转录组数据筛选出的SSR位点设计出54对SSR引物,对其9个不同地理种群进行验证,有16对引物能较好扩增出目的片段。桑迪等[14]基于意大利蝗Calliptamus italicus转录组数据设计出6对可用于扩增10个不同地理种群目的片段的SSR引物。目前,有关黄地老虎的研究,主要集中在人工饲养、生物学特性、预测预报和防治技术等方面[17-19],而对其种群遗传结构方面的研究甚少。Wu等[20]仅对黄地老虎线粒体基因组进行简单分析,并未利用线粒体基因对其系统发育和遗传结构进行研究。【本研究切入点】对害虫种群遗传结构进行研究,不仅能够分析出害虫在我国现有地理分布格局的形成机制,而且能够了解该虫在不同地理种群间的遗传分化与基因流,结合该虫在我国的迁飞与气象资料,还能够推测该虫在我国的迁飞规律及各发生危害区间的虫源关系。黄地老虎作为一种重要的迁飞性农业害虫,缺少对其种群遗传结构方面的研究,明确黄地老虎SSR和SNP位点信息可为研究该虫种群遗传结构提供数据支撑。【拟解决的关键问题】本研究通过对黄地老虎转录组进行分析,对其SSR和SNP位点的组成和特征进行研究,初步建立黄地老虎的分子标记,为今后研究黄地老虎的种群遗传结构奠定基础。
1 材料与方法 1.1 供试虫源及RNA提取黄地老虎成虫采自山东省烟台市蓬莱区北隍城乡长岛试验基地(站)(38 °23.20´N,120°54.50´E)。选取鳞片完整且活力充沛的个体放在1.5 mL的离心管内,迅速置于液氮中致死,将样品存放于-80 ℃超低温冰箱内直至使用。
在已灭菌的研钵中加入液氮预冷,将黄地老虎样品倒入预冷的研钵中,迅速研磨直至研磨成粉末状,期间不断补充液氮。将约80 mg粉末状样品转移至1.5 mL的离心管内,加入1 mL TransZol Up(北京全式金公司)溶液后,采用RNA提取试剂盒(TransZol Up Plus RNA Kit,北京全式金公司)提取总RNA。对总RNA质量采用1%琼脂糖凝胶电泳进行检测;Nanodrop分光光度计(IMPLEN,CA,USA)对总RNA浓度和纯度进行检测;利用Agilent 2 100(Agilent Technologies,CA,USA)检测RNA的完整性。
1.2 转录组测序及序列拼接将检测质量合格的总RNA,委托深圳市恒创基因科技有限公司用Illumina HiSeq 2500进行测序,共3个生物学重复,测序深度为10 Gb。对测序获得的原始数据进行过滤,去除包含接头的序列及低质量序列,使用Trinity软件对去重复的clean reads进行De novo组装,获得Unigene序列。
1.3 SSR和SNP信息分析对测序获得的黄地老虎转录组数据,使用MicroSAtellite(MISA)软件对Unigene进行检测,得到SSR位点信息[21]。此外,利用GATK软件对黄地老虎SNP信息进行分析[22]。
2 结果与分析 2.1 转录组数据分析黄地老虎转录组经Illumina测序后,共得到74 067 260条原始数据;数据过滤后,得到73 724 516条clean reads,其中,Q20为93.52%,Q30为85.81%,表明测序质量较好。组装后共得到66 469条Unigene序列,序列长度主要分布于200~2 000 bp之间,占总序列的89.88%,平均长度868 bp。随着Unigene序列长度的逐渐增加,所含序列数量呈阶梯式下降(图 1),该结果同生物的序列长度分布规律相一致,说明该转录组组装质量较好。

|
| 图 1 Unigene序列长度分布 Fig. 1 Unigene Distribution of length in Unigene distribution |
2.2 SSR位点分析
对获得的66 469条Unigene序列利用MISA软件进行搜索,共得到SSR位点4 438个。分布于4 048条Unigene序列上,占总序列的6.09%;其中包含多个微卫星位点的序列有345条,复合型SSR有125条。
对SSR位点的核苷酸重复类型进行分类,共得到6种重复类型,各重复类型间所包含的SSR位点的数目相差较大;其中,重复类型最多的是单核苷酸重复(2 429个),占总位点的54.73%;六核苷酸重复类型的位点数最少,占比0.95%(表 1)。黄地老虎SSR总长度为67 966 bp,其中单核苷酸重复序列总长度最长,为37 215 bp,平均每1 552 bp出现1个单核苷酸重复,其平均长度为15.32 bp;六核苷酸重复SSR位点总长度最短,为1 056 bp,其平均长度为25.14 bp。6种重复类型序列总长度为67 966 bp,平均每850 bp出现1个重复序列,其平均长度为15.31 bp(表 1)。
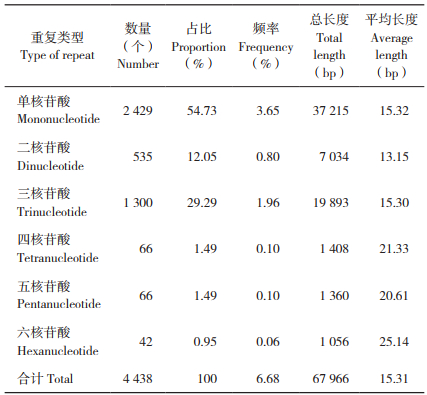
黄地老虎转录组SSR位点信息中共发现82种重复基元,其中单核苷酸重复基元种类最少,为2种;六核苷酸重复基元种类最多,为25种。A/T为单核苷酸重复基元中的优势基元,共有SSR位点2 356个,分别占单核苷酸重复基元和总SSR位点的96.99%和53.09%。二核苷酸重复基元中的优势基元为AC/GT和CG/CG;三核苷酸重复基元中的优势基元是ATC/ATG和CCG/CGG;四核苷酸重复基元中的优势基元是AAAT/ATTT和AAAG/CTTT;五核苷酸重复基元中的优势基元是AAATC/ATTTG和AATAG/ATTCT;六核苷酸重复基元中的优势基元是CCCGCG/CGCGGG(表 2)。
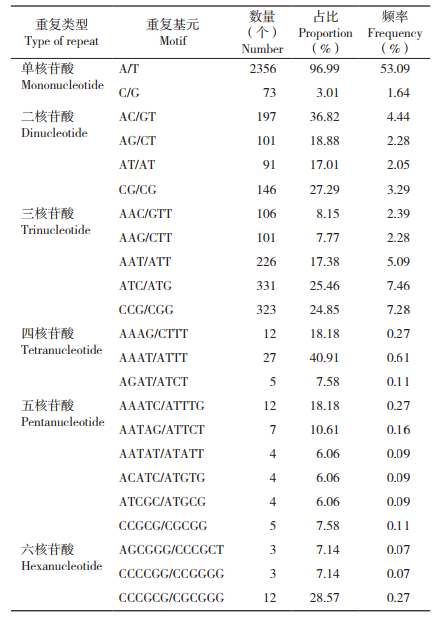
黄地老虎转录组中SSR基元重复次数在不同基元类型间存在较大差异,包含4~24次重复;且重复次数随着基元核苷酸数的增加而呈下降趋势。单核苷酸重复基元的重复次数类型最多,共有13种重复单元长度,为12~24,其中12次重复所占比例最高,占单核苷酸SSR位点的32.24%。六核苷酸重复基元形成3种重复单元长度,其中4次重复所占比例最高,占六核苷酸SSR位点的88.10%(表 3)。
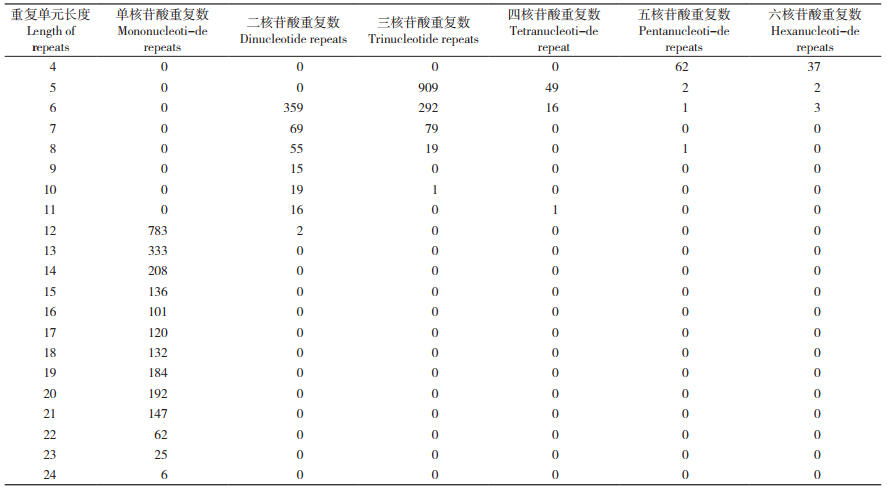
对黄地老虎转录组SSR位点序列长度进行分析,SSR位点序列长度分布在12~127 bp之间。SSR位点序列长度在12~19 bp的SSR位点有3 493个,其中,SSR长度为12 bp的位点数量最多,占总位点的25.39%;长度为15 bp的SSR位点数量次之,占总位点的22.49%。SSR位点序列长度大于等于20 bp的SSR位点有820个,占总位点的19.01%(图 2)。

|
| 图 2 SSR位点序列长度分布 Fig. 2 Distribution of length in SSR loci sequences |
2.3 SNP位点分析
黄地老虎转录组Unigene序列共包含SNP位点371 148个,包括237 619个转换类型和133 529个颠换类型;转换类型(64.02%)显著大于颠换类型(35.98%)。C/T在所有变异类型中所占比率最高,占总量的18.61%;G/A位居其次(18.28%),且这两种变异均属于转换类型。在颠换类型中,A/T所占比例最高,占总量的6.93%;G/C所占比例最低,占总量的3.29%(图 3)。

|
| 图 3 SNP变异类型统计结果 Fig. 3 Statistical results of variation types of SNP |
3 讨论
本研究基于黄地老虎转录组测序结果,对其SSR及SNP位点信息进行分析。黄地老虎转录组数据共获得66 469条Unigene序列,共搜索到SNP位点371 148个,平均每155 bp出现一个SNP位点。黄地老虎转录组数据中SNP变异的转换位点有237 619个,颠换位点有133 529个,说明其SNP变异类型以转换类型为主,该结果与东方蝼蛄Gryllotalpa orientialis[23]、椰心叶甲啮小蜂Tetrastichus brontispae[24]的研究结果一致。本研究中转换位点是颠换位点的1.78倍,小于理论值(转换/颠换=1∶2),说明碱基的突变可能不是随机的,应该与进化过程中的选择机制相关[25]。在转换类型中,C/T所占的比率最高,可能是由于甲基化的C进行脱氨后就可以变成T有关[26]。
黄地老虎转录组数据库中共搜索到SSR位点4 438个,分布于4 048条Unigene序列上,发生频率为6.09%,比已报道的部分昆虫如扶桑绵粉蚧Phenacoccus solenopsis(5.79%)[27]、桔小实蝇Bactrocera dorsalis(4.23%)[28]、麦红吸浆虫Sitodiplosis mosellana(3.49%)[29]的发生频率高;而比东方黏虫Mythimna separate(9.85%)[30]、荔枝蒂蛀虫Conopomorpha sinensis(15.25%)[31]、意大利蝗(17.58%)[14]和沙棘木蠹蛾Eogystia hippophaecolus(35.14%)[32]等昆虫的发生频率低,不同昆虫间SSR发生频率差异较大,造成该结果的原因可能与物种自身进化有关,也可能同用于建库的总RNA质量、测序深度以及数据分析时参数设置有关[15]。
黄地老虎转录组中的SSR包括6种重复类型,其中单碱基重复是含量最高的重复类型,该结果与东方黏虫[30]、荔枝蒂蛀虫[31]、椰心叶甲啮小蜂[24]、梨小食心虫Grapholitha molesta[16]等的研究结果相一致。其次,三碱基重复的含量也较高,在桔小实蝇[28]、二点委夜蛾Athetis lepigone[33]、黏虫Mythimna separata[34]等昆虫中,三碱基重复是含量最高的重复类型,导致三碱基重复含量较高的原因可能在于其较其他重复类型更加稳定,每3个核苷酸翻译1个氨基酸,为保证蛋白质结构与功能不产生变化,使得其极少产生移码突变[35]。
前人研究表明,由于C易于甲基化突变成T,推测在动植物体内GC/CG的含量会很低,有些甚至不含该重复基元[36-37]。然而,本研究中CG/CG含量是除AC/GT外含量最高的二碱基重复,该结果同一些鳞翅目昆虫如二点委夜蛾[33]、东方黏虫[30]和细梢小卷蛾Rhyacionia leptotubula[38]的研究结果相一致,可能是由于鳞翅目昆虫自身的进化导致其含量较高,需要进一步研究。
SSR多态性是判断序列能否作为分子标记的重要依据,而SSR序列长度是影响SSR多态性的主要因素,且序列长度与多态性成正比[36, 39]。研究结果表明,SSR多态性极低的序列其长度小于12 bp,SSR多态性中等的序列其长度在12~20 bp之间,而SSR多态性较高的序列其长度大于等于20 bp[39]。本研究中,黄地老虎SSR位点序列长度在12~19 bp的SSR位点有3 493个,属于SSR中度多态性位点;大于等于20 bp的SSR位点有820个,属于SSR高度多态性位点。总体来说,黄地老虎具有相对较高的遗传多态性。
4 结论本研究基于黄地老虎转录组数据库,对黄地老虎SSR和SNP位点信息进行分析,结果显示该虫转录组中包含SNP位点371 148个和SSR位点4 438个;SNP以转换类型(64.02%)为主;SSR位点共存在6种重复类型,以5次重复(21.67%)为主,其序列长度为12~127 bp,包含3 493个中等多态性位点和820个高度多态性位点。研究结果表明该虫SNP和SSR位点数较多,SSR重复类型丰富,且序列长度较长,具有较高的遗传多态性,可为研究黄地老虎遗传多样性提供充分的遗传信息,用于黄地老虎种群遗传分化的研究。
| [1] |
LEMIC D, DRMIĆ Z, BAŽOK R. Population dynamics of noctuid moths and damage forecasting in sugar beet[J]. Agricultural and Forest Entomology, 2016, 18(2): 128-136. DOI:10.1111/afe.12145 |
| [2] |
NOWINSZKY L, KISS M, PUSKSÁk J, BARTA A. Light-trap catch of turnip moth (Agrotis segetum Denis et Schiffermüller, 1775) in connection with the night sky polarization phenomena[J]. Global Journal of Research and Review, 2017, 4(2): 22-31. DOI:10.21767/2393-8854.100022 |
| [3] |
GUO J L, FU X W, WU X, ZHAO X C, WU K M. Annual migration of Agrotis segetum(Lepidoptera: Noctuidae): observed on a small isolated island in northern China[J]. PLoS One, 2015, 10(6): e0131639. DOI:10.1371/journal.pone.0131639 |
| [4] |
CHANG H, GUO J L, FU X W, LIU Y Q, WYCKHUYS K A G, HOU Y M, WU K M. Molecular-assisted pollen grain analysis reveals spatiotemporal origin of long-distance migrants of a Noctuid moth[J]. International Journal of Molecular Sciences, 2018, 19(2): 567-572. DOI:10.3390/ijms19020567 |
| [5] |
DUAN X L, PENG X, QIAO X F, CHEN M H. Life cycle andpopulation genetics of bird cherry-oat aphids Rhopalosiphum padi in China: an important pest on wheat crops[J]. Journal of Pest Science, 2017, 90(1): 103-116. DOI:10.1007/s10340-016-0752-9 |
| [6] |
陈静, 何吉祥, 黄龙, 吴本丽, 孙和权, 宋光同, 陈贵生, 王晓健. 3个中华鳖群体遗传多样性的微卫星标记分析[J]. 广东农业科学, 2017, 44(7): 141-146. DOI:10.16768/j.issn.1004-874X.2017.07.022 CHEN J, HE J X, HUANG L, WU B L, SUN H Q, SONG G T, CHEN G S, WANG X J. Microsatellite marker analysis of genetic diversity in three populations of Trionyx sinensis[J]. Guangdong Agricultural Sciences, 2017, 44(7): 141-146. DOI:10.16768/j.issn.1004-874X.2017.07.022 |
| [7] |
徐放, 朱报著, 潘文, 朱政财, 钟乃盛, 李文业, 赵强民. 基于SSR标记的广东含笑等11个含笑属物种亲缘关系研究[J]. 广东农业科学, 2021, 48(1): 87-93. DOI:10.16768/j.issn.1004-874X.2020.07.011 XU F, ZHU B Z, PAN W M, ZHU C Z, ZHONG N S, LI W Y, ZHAO Q M. Study on the genetic relationship of 11 michelia species based on SSR markers[J]. Guangdong Agricultural Sciences, 2021, 48(1): 87-93. DOI:10.16768/j.issn.1004-874X.2020.07.011 |
| [8] |
李清, 罗永坚, 吴柔贤, 贾俊婷, 张文虎, 宋松泉, 刘军. 广东省大豆种质资源遗传多样性分析及DNA分子身份证构建[J]. 广东农业科学, 2020, 47(12): 221-228. DOI:10.16768/j.issn.1004-874X.2020.12.023 LI Q, LUO Y Q, WU R X, JIA J T, ZHANG W H, SONG S Q, LIU J. Analysis on genetic diversity and construction of DNA molecular identity card of soybean germplasm resources in Guangdong province[J]. Guangdong Agricultural Sciences, 2020, 47(12): 221-228. DOI:10.16768/j.issn.1004-874X.2020.12.023 |
| [9] |
Campbell N R, Narum S R. Quantitative PCR assessment of microsatellite and SNP genotyping with variable quality DNA extracts[J]. Conservation Genetics, 2009, 10(3): 779-784. DOI:10.1007/s10592-008-9661-7 |
| [10] |
王晨, 马宁, 郭春和, 袁仁强, 曾检华, 宋德清, 张惠文, 陈瑶生, 刘小红. 基于SNP芯片分析的蓝塘猪遗传群体结构[J]. 广东农业科学, 2018, 45(6): 110-115. DOI:10.16768/j.issn.1004-874X.2018.06.018 WANG C, MA N, GUO C H, YUAN R Q, ZENG J H, SONG D Q, ZHANG H W, CHEN Y S, LIU X H. Population structure of Lantang pig evaluated using SNP chip[J]. Guangdong Agricultural Sciences, 2018, 45(6): 110-115. DOI:10.16768/j.issn.1004-874X.2018.06.018 |
| [11] |
YANG J P, LI X H, LI Y F, ZHU S L, CHEN W T, LI J. Isolation and characterization of 30 SNP markers in Guangdong bream (Megalobrama terminalis)by next-generation sequencing[J]. Conservation Genetics Resources, 2020, 12: 399-402. DOI:10.1007/s12686-020-01131-1 |
| [12] |
GAO X G, Bao X B, SUN W, LI Y F, LIU Z Y, LIU W D. Isolation and characterization of 38 SNP markers for the black rockfish, Sebastes schlegelii by next-generation sequencing[J]. Conservation Genetics Resources, 2021, 1-4. DOI:10.1007/S12686-021-01226-3 |
| [13] |
黎东海, 赵萍. 基于转录组数据的齿缘刺猎蝽微卫星分子标记开发[J]. 昆虫学报, 2019, 62(6): 694-702. DOI:10.16380/j.kcxb.2019.06.005 LI D H, ZHAO P. Development of microsatellite markers based on the transcriptome data of Sclomina erinacea(Heteroptera: Reduviidae)[J]. Acta Entomologica Sinica, 2019, 62(6): 694-702. DOI:10.16380/j.kcxb.2019.06.005 |
| [14] |
桑迪, 徐叶, 王伟亮, 向敏, 季荣, 王晗. 基于转录组数据的意大利蝗微卫星位点分析与分子标记开发[J]. 应用昆虫学报, 2020, 57(3): 658-666. DOI:10.7679/j.issn.2095-1353.2020.066 SANG D, XU Y, WANG W L, XIANG M, JI R, WANG H. Analysis of microsatellite loci from Calliptamus italicus(Orthopera: Acrididae)based on a transcriptome dataset[J]. Chinese Journal of Applied Entomology, 2020, 57(3): 658-666. DOI:10.7679/j.issn.2095-1353.2020.066 |
| [15] |
DENG K P, DENG R J, FAN J X, CHEN E F. Transcriptome analysis and development of simple sequence repeat(SSR)markers in Zingiber striolatum Diels[J]. Physiology and Molecular Biology of Plants, 2018, 24(1): 125-134. DOI:10.1007/s12298-017-0485-0 |
| [16] |
冷春蒙, 李引, 胡迪, 仵均祥, 李怡萍. 梨小食心虫幼虫中肠转录组及SSR分子标记分析[J]. 昆虫学报, 2018, 61(11): 1272-1283. DOI:10.16380/j.kcxb.2018.11.004 LENG C M, LI Y, HU D, WU J X, LI Y P. Analysis of the larval midgut transcriptome and SSR markers in Grapholitha molesta (Lepidoptera: Tortricidae)[J]. Acta Entomologica Sinica, 2018, 61(11): 1272-1283. DOI:10.16380/j.kcxb.2018.11.004 |
| [17] |
陈婧, 刘蓉, 梁虎军, 罗树凯, 罗凤君. 新疆阿拉尔垦区黄地老虎种群监测与发生特点分析[J]. 中国棉花, 2021, 48(7): 26-28, 36. DOI:10.11963/cc20210066 CHEN J, LIU R, LIANG H J, LUO S K, LUO F J. Population monitoring and occurrence characteristics of Agrotis segetum Schiff. in Aral Reclamation Area of Xinjiang[J]. China Cotton, 2021, 48(7): 26-28, 36. DOI:10.11963/cc20210066 |
| [18] |
郭江龙, 付晓伟, 赵新成, 吴孔明. 黄地老虎飞行能力研究[J]. 环境昆虫学报, 2016, 38(5): 888-895. DOI:10.3969/j.issn.1674-0858.-2016.05.3 GUO J L, FU X W, ZHAO X C, WU K M. Preliminary study on the flight capacity of Agrotis segetum(Lepidoptera: Noctuidae)[J]. Journal of Environmental Entomology, 2016, 38(5): 888-895. DOI:10.3969/j.issn.1674-0858.-2016.05.3 |
| [19] |
李琳, 修春丽, 路伟, 陆宴辉. 黄地老虎成虫对15种植物挥发物的电生理和行为反应[J]. 新疆农业科学, 2020, 57(11): 2020-2027. DOI:10.6048/j.issn.1001-4330.2020.11.008 LI L, XIU C L, LU W, LU Y H. Electrophysiological and behavioral responses of Agrotis Segetum adults to 15 plant volatiles[J]. Xinjiang Agricultural Sciences, 2020, 57(11): 2020-2027. DOI:10.6048/j.issn.1001-4330.2020.11.008 |
| [20] |
WU Q L, CUI W X, DU B Z, GU Y, WEI S J. The complete mitogenome of the turnip moth Agrotis segetum(Lepidoptera: Noctuidae)[J]. Mitochondrial DNA, 2014, 25(5): 345-347. DOI:10.3109/19401736.2013.800496 |
| [21] |
THIEL T, MICHALEK W, VARSHNEY R K, GRANER A. Exploiting EST databases for the development and characterization of genederived SSR-markers in barley (Hordeum vulgare L.)[J]. Theoretical and Applied Genetics, 2003, 106(3): 411-422. DOI:10.1007/s00122-002-1031-0 |
| [22] |
MCKENNA A, HANNA M, BANKS E, SIVACHENKO A, CIBULSKIS K, KERNYTSKY A, GARIMELLA K, ALTSHULER D, GABRIEL S, DALY M, DEPRISTO M A. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data[J]. Genome Research, 2010, 20(9): 1297-303. DOI:10.1101/gr.107524.110 |
| [23] |
李霞. 东方蝼蛄(Gryllotalpa orientialis)线粒体基因组及转录组研究[D]. 西安: 陕西师范大学, 2014. LI X. The study of mitochondrial genome and transcriptome of Gryllotalpa orientialis[D]. Xian: Shaanxi Normal University, 2014. |
| [24] |
刘华伟, 李朝绪, 李芬, 吕朝军, 吴少英, 覃伟权. 基于转录组测序的椰心叶甲啮小蜂SSR、SNP和InDel位点分析[J]. 热带作物学报, 2021, 42(10): 2828-2833. LIU H W, LI C X, LI F, LV C J, WU S Y, QIN W Q. SSR, SNP and In Del analysis based on Tetrastichus brontispae transcriptome[J]. Chinese Journal of Tropical Crops, 2021, 42(10): 2828-2833. |
| [25] |
ZHAO H, LI Q Z, LI J, ZENG C Q, HU S N, YU J. The study of neighboring nucleotide composition and transition/transversion bias [J]. Science in China Series C: Life Sciences, 2006, 49(4): 395-402. DOI:cnki:sun:jcxg.0.2006-04-011.
|
| [26] |
GARG K, GREEN P, NICKERSON D A. Identification of candidate coding region single nucleotide polymorphisms in 165 human genes using assembled expressed sequence tags[J]. Genome Research, 1999, 9(11): 1087-1092. DOI:10.1101/gr.9.11.1087 |
| [27] |
罗梅, 张鹤, 宾淑英, 林进添. 基于转录组数据高通量发掘扶桑绵粉蚧微卫星引物[J]. 昆虫学报, 2014, 57(4): 395-400. DOI:10.16380/j.kcxb.2014.04.005 LUO M, ZHANG H, BIN S Y, LIN J T. High-throughput discovery of SSR genetic markers in the mealybug, Phenacoccus solenopsis (Hemiptera: Pseudococcidae), from its transcriptome database[J]. Acta Entomologica Sinica, 2014, 57(4): 395-400. DOI:10.16380/j.kcxb.2014.04.005 |
| [28] |
魏丹丹, 石俊霞, 张夏瑄, 陈世春, 魏冬, 王进军. 基于转录组数据的桔小实蝇微卫星位点信息分析[J]. 应用生态学报, 2014, 25(6): 1799-1805. DOI:10.13287/j.1001-9332.20140409.022 WEI D D, SHI J X, ZHANG X X, CHEN S C, WEI D, WANG J J. Analysis of microsatellite loci from Bactrocera dorsalis based on transcriptome dataset[J]. Chinese Journal of Applied Ecology, 2014, 25(6): 1799-1805. DOI:10.13287/j.1001-9332.20140409.022 |
| [29] |
段云, 吴仁海, 罗礼智, 武予清, 蒋月丽, 苗进, 巩中军. 麦红吸浆虫唾腺EST-SSRs的信息分析及分子标记筛选[J]. 昆虫学报, 2011, 54(10): 1147-1154. DOI:10.16380/j.kcxb.2011.10.003 DUAN Y, WU R H, LUO L Z, DUAN Y Q, JIANG Y L, MIAO J, GONG Z J. Characterization of SSRs from the ESTs in the wheat midge, Sitodiplosis mosellana(Gehin) (Diptera: Cecidomyiidae)[J]. Acta Entomologica Sinica, 2011, 54(10): 1147-1154. DOI:10.16380/j.kcxb.2011.10.003 |
| [30] |
李微, 张蕾, 程云霞, 罗礼智, 江幸福. 应用转录组测序高通量发掘东方粘虫SSR标记[J]. 植物保护学报, 2017, 44(3): 377-384. DOI:10.13802/j.cnki.zwbhxb.2017.2016058 LI W, ZHANG L, CHENG Y X, LUO L Z, JIANG X F. High-throughput discovery of microsatellite markers based on transcriptome sequencing in the oriental armyworm, Mythimna separate(Walker)[J]. Journal of Plant Protection, 2017, 44(3): 377-384. DOI:10.13802/j.cnki.zwbhxb.2017.2016058 |
| [31] |
孟翔, 胡俊杰, 李艳华, 欧阳革成. 基于转录组数据的荔枝蒂蛀虫SSR位点信息分析[J]. 环境昆虫学报, 2017, 39(6): 1219-1224. DOI:10.3969/j.issn.1674-0858 MENG X, HU J J, LI Y H, OUYANG G C. Analysis of SSR loci in transcriptome database of Conopomorpha sinensis Bradley (Lepidoptera: Gracilariidae)[J]. Journal of Environmental Entomology, 2017, 39(6): 1219-1224. DOI:10.3969/j.issn.1674-0858 |
| [32] |
崔明明, 陶静, 宗世祥. 基于转录组的沙棘木蠹蛾简单重复序列特征分析[J]. 环境昆虫学报, 2017, 39(3): 605-610. DOI:10.3969/j.issn.1674-0858.2017.03.15 CUI M M, TAO J, ZONG S X. Feature analysis of simple sequence repeats in Eogystia hippophaecolus transcriptome[J]. Journal of Environmental Entomology, 2017, 39(3): 605-610. DOI:10.3969/j.issn.1674-0858.2017.03.15 |
| [33] |
LI L T, ZHU Y B, MA J F, LI Z Y, DONG Z P. An analysis of the Athetis lepigone transcriptome from four developmental stages[J]. PLOS ONE, 2013, 8(9): e73911. DOI:10.1371/journal.pone.0073911 |
| [34] |
胡艳华, 李敏, 张虎芳, 李生才, 王青, 赵惠玲. 粘虫转录组中SSR位点的信息分析[J]. 山西农业大学学报(自然科学版), 2015, 35(5): 484-489. DOI:10.13842/j.cnki.issn1671-8151.2015.05.007 HU Y H, LI M, ZHANG H F, LI S C, WANG Q, ZHAO H L. The information analysis of SSR loci in the Mythimna separata (Walker) transcriptome[J]. Journal of Shanxi Agricultural University (Natural Science Edition), 2015, 35(5): 484-489. DOI:10.13842/j.cnki.issn1671-8151.2015.05.007 |
| [35] |
WANG B, EKBLOM R, CASTOE T A, JONES E P, KOZMA R, BONGCAM-RUDLOFF E, POLLOCK D D, HÖGLUND J. Transcriptome sequencing ofblack grouse (Tetrao tetrix) for immune gene discovery andmicrosatellite development[J]. Open Biology, 2012, 2(4): 120054. DOI:10.1098/rsob.120054 |
| [36] |
MRGLÉCZ E, NÈVE G, BIFFIN E, GARDNER M G. Breakdown of phylogenetic signal: A survey of microsatellite densities in 454 shotgun sequences from 154 non model eukaryote species[J]. PLOS ONE, 2012, 7(7): e40861. DOI:10.1371/journal.pone.0040861 |
| [37] |
YOON J M. Genetic variations between hairtail (Trichiurus lepturus) populations from Korea and China[J]. Development & Reproduction, 2013, 17(4): 363-367. DOI:10.12717/DR.2013.17.4.363 |
| [38] |
ZHU J Y, LI Y H, YANG S, LI Q W. De novo Assembly and characterization of the global transcriptome for Rhyacionia leptotubula using illumina paired-end sequencing[J]. PLOS ONE, 2013, 8(11): e81096. DOI:10.1371/journal.pone.0081096 |
| [39] |
TEMNYKH S, DECLERCK G, LUKASHOVA A, LIPOVICH L, CARTINHOUR S, MCCOUCH S. Computational and experimental analysis of microsatellites in rice(Oryza sativa L.): frequency, length variation, transposon associations, and genetic marker potential[J]. Genome Research, 2001, 11(8): 1441-1452. DOI:10.1101/gr.184001 |
(责任编辑 杨贤智)