 2021, Vol. 48
2021, Vol. 48文章信息
引用本文 |

基金项目
- 肇庆市科技计划项目(2020N13004);广东省农业科学院中青年学科带头人培养项目(R2019PYJX003);广东省农业科学院创新基金-产业专项(202148)
作者简介
-
梅瑜(1984—),女,博士,助理研究员,研究方向为药用植物栽培育种,E-mail:meiyu@gdaas.cn
梅瑜,博士,助理研究员,药用植物资源工程专业,主要从事华南特色作物(南药)的育种与栽培研究工作。主持和参加科技项目10多项,对接“广东省省级现代农业产业园”科技服务团队5个。在国内外学术期刊发表科技论文20余篇,授权专利5项,软件著作权7项.
通讯作者
- 王继华(1979—),男,博士,研究员,研究方向为药用植物栽培育种,E-mail:wangjihua@gdaas.cn.
文章历史
- 收稿日期:2021-10-31



【研究意义】药用植物遗传资源是优良品种选育的基础。近年来随着市场需求的增长,导致野生资源枯竭、驯化栽培品种品质参差不齐。利用高通量测序技术能够精准评价药用资源,解析其药用活性的合成途径和调控机制,有利于药用资源的保护和利用。【前人研究进展】艾蒿(Artemisia argyi),是菊科蒿属植物,多年生草本或略呈半灌木状,具浓烈香气,全草入药,有温经、祛湿、散寒、止血、安胎、通经活络等功效。艾蒿中的挥发油类[1]、黄酮类、萜类、生物碱类、多糖类[2]等天然化学产物和大量微量元素[3],具有镇咳平喘、杀菌消炎、益气活血、温经活络、抗氧化等功效[4],艾灸具有增强免疫力和抗炎作用。其黄酮的主要成分包括甲基黄酮类、槲皮素和柚皮素等[5]。胡倩等[6]利用AB-8型大孔树脂分离纯化了艾叶总黄酮提取物(TFEFAA),采用HPLC分析获得TFEFAA的主要成分为棕矢车菊素和异泽兰黄素,虽然其质量分数分别只有0.6% 和0.20%,但能显著增加SOD酶、GSHPx酶活力,且降低MDA含量,能有效清除自由基和提高机体抗氧化能力。艾蒿还可作为天然的杀虫剂、植物染料和饲料添加剂。艾蒿药用功能显著、价格低廉,因此在医药、农业、工业等领域被广泛利用,也受到学者们青睐。艾蒿品种较多,分布较为广泛,药用历史悠久。梅全喜[7]记载艾蒿20多种,红足蒿(红脚艾)是其中之一。三元宫碑文记载,1600年前葛洪、鲍菇夫妇在罗浮山用红脚艾为当地百姓治病,因此罗浮山红脚艾在岭南地区较有代表性,又称“红艾”“鲍姑艾”,别名神艾,与北艾、蕲艾同属于菊科蒿属艾草种植物。岭南红脚艾为菊科红足蒿(Artemisia rubripes Nakai),生长在阳光充足的田野,茎枝呈紫红色,多有横卧地下根状茎伴匍匐生长,植株最高1 m左右,香气醇厚,基本为野生。红脚艾常用于艾灸,是临床应用艾叶的来源之一。其蛋白质中含有16种常见氨基酸,7种为必需氨基酸,必需氨基酸占总氨基酸的40.97%,功能性氨基酸占总氨基酸的53.6%,且叶小绒少,味略苦(其他品系艾草较苦) [8],因此广东当地人常将红脚艾用作主要的食材原料之一。【本研究切入点】目前,艾蒿的研究多集中于化学成分的分离鉴定、提取工艺优化,以及药理作用研究,而红脚艾相关研究较少,尤其对红脚艾组学研究和次生代谢产物合成途径解析及化合物合成关键基因鉴定等未见报道。【拟解决的关键问题】由于红脚艾基因组信息缺乏,本研究利用高通量测序平台Illumina HiSeq 2500,选择红脚艾不同组织进行混合DNA池测序,通过Trinity软件de novo组装获得红脚艾转录组Unigene库,并基于公共数据库对Unigene进行功能注释,为后续的次生代谢产物合成途径研究、相关功能基因挖掘及验证提供基础数据。
1 材料与方法 1.1 试验材料供试材料红脚艾蒿(A. rubripes Nakai)种植于广东省农业科学院作物研究所南药资源圃,由王继华研究员鉴定。选择长势基本一致的健康植株,采摘叶片,用锡箔纸包裹迅速置于液氮中。
1.2 红脚艾RNA的提取红脚艾总RNA采用生工生物Total RNA Extractor kit(B511311-0025)进行提取,用1% 琼脂糖电泳检测总RNA的完整性,用Qubit® 2.0荧光计Life Tech(Invitrogen)对总RNA进行定量。
1.3 转录组测序与功能注释红脚艾RNA样品检测合格后进行转录组测序。利用Illumina®NEBNext®UltraTM RNA Library Prep Kit制备测序文库,之后使用Agilent Bioanalyzer 2100评估文库质量;构建合格文库后,利用Illumina Hiseq 2500平台进行测序,获得双端测序数据;过滤原始数据中的接头、低质量(reads ≤ 35 nt)和ploy-N碱基的序列,获得clean data,计算GC、Q20、Q30含量及序列重复水平;采用Trinity软件进行de novo拼接组装,用RSeQC软件去除冗余序列获得Unigene, 并评估基因表达水平[9-10]。利用BLAST软件[11],基于NR、GO、COG、KOG、KEGG等数据库[12-18]对组装的Unigene进行功能注释。
1.4 SSR鉴定及分析使用MISA微卫星识别工具鉴定简单重复序列(Simple Sequence Repeat,SSR)并进行分析。
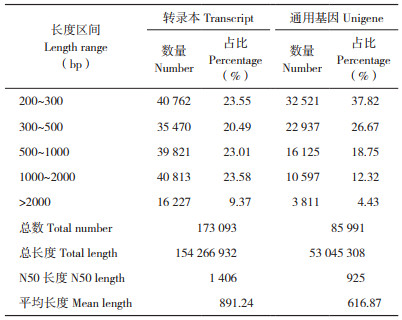
2 结果与分析 2.1 红脚艾转录组测序结果利用Illumina Hiseq 2500平台对红脚艾RNA样品进行转录组测序后,去掉含有接头和低质量的序列,共得到7.24 Gb clean data,24 126 043条reads,GC含量为44.67%,各样品Q30碱基百分比均不小于92.57%,说明文库构建质量良好,试验数据可靠。经de novo组装,共得到173 093个转录本,平均长度为891.24 bp,N50为1 406 bp,序列长度大于500 bp的有96 861个,占总序列数目的55.96%;转录本去冗余后得到85 991个Unigene,平均长度为616.87 bp,N50为925 bp,其中30 533个(35.51%)序列长度大于500 bp,有14 408个(16.75%)序列长度在1 000 bp以上(表 1)。
|
2.2 Unigene功能注释
通过BLAST软件将获得的85 991个Unigene序列与不同功能数据库进行比对,共注释到47 216个(54.91%)Unigene(表 2)。其中,NR数据库(40 802个,47.45%)、KOG数据库(26 171个,30.43%)、GO数据库(23 203个,26.98%) 和KEGG数据库(16 846个,19.59%),COG数据库注释到的Unigene数量最少(12 810个,14.9%)。

2.2.1 NR功能注释 NR数据库注释结果显示,红脚艾与向日葵(Helianthus annuus)的单基因匹配率(13 552个,15.76%)最高,其次是莴苣(Lactuca sativa,9 187个,10.68%)、洋蓟(Cynara cardunculus var. scolymus,6 609个,7.69%),与菊花(Chrysanthemum × morifolium)、莫氏黑粉菌(Moesziomyces antarcticus T-34)、青稞(Hordeum vulgare subsp. Vulgare)的单基因匹配均小于400个。可见,红脚艾与菊目菊科物种的相似度高。
2.2.2 GO功能注释 GO注释结果(图 1) 显示,红脚艾有23 203个Unigene被归为生物过程(43 283个,50.33%)、细胞组成(47 818个,55.79%) 和分子功能(27 375个,31.83%) 三大类共51个功能分类。在分子功能中,催化活性(12 156个,14.14%) 和结合(11 434个,13.30%)注释到的Unigene较多;在细胞组分中,细胞(9 631个,11.20%) 和细胞部分(9 607个,11.17%)注释到的Unigene较多;生物过程中,代谢过程(11 847个,13.78%)和分子过程(10 948个,12.73%)注释最丰富。

|
| 图 1 红脚艾Unigene的GO功能分类 Fig. 1 Classification of GO function of Artemisia rubripes Nakai Unigene |
2.2.3 KEGG代谢通路分析 从KEGG数据库注释结果来看,16 846个Unigene参与到130条代谢途径中。其中核糖体途径注释的基因较丰富(824个,4.89%),其次是碳代谢(604个,3.59%)、剪切体(503个,2.99%)、内质网中蛋白质加工(474个,2.81%)和氨基酸生物合成(466个,2.77%) 等。参与类黄酮生物合成的基因有49个,仅占0.29%。萜类作为自然界中结构多、种类多的天然化合物,其生物合成分为前体合成、骨架构建和后修饰3部分。本研究获得萜类骨架合成过程Unigene 105个,参与直接前体GPP(Geranyl-PP)、IPP(Isopentenyl-PP)及DMAPP(Dimethylallyl-PP) 的形成,参与单萜生物合成相关的Unigene有13个,参与二萜生物合成的Unigene有27个,参与倍半萜和三萜生物合成相关的Unigene有14个,萜类经细胞色素P450等酶修饰后形成化合物,如二萜类化合物对肿瘤有一定治疗作用,二萜类激素Gas对植物发育具有调控作用,三萜皂苷具有很高的药用价值等。
2.2.4 KOG功能分类 KOG数据库分析结果(图 2)显示,红脚艾26 171个Unigene被注释到25个KOG功能分类中。其中,一般功能预测的基因最多,共有5 458个,占20.86%;其次是信号传导机制相关基因(2 976个,11.37%),翻译后修饰、蛋白质周转、伴侣基因(2 862个,10.94%),翻译、核糖体结构与生物发生相关基因(1 690个,6.46%),转录相关基因(1 527个,5.83%),细胞内运输、分泌和囊泡转运相关基因(1 374个,5.25%),碳水化合物运输和代谢相关基因(1 319个,5.04%),与次生代谢物生物合成、运输和分解代谢相关基因(1 164个,4.45%) 等,细胞运动注释到的基因最少,仅18个,占0.07%;未知功能的基因有1 491个,需在后期挖掘其功能。

|
| 图 2 红脚艾Unigene的KOG功能分类 Fig. 2 Classification of KOG function of Artemisia rubripes Nakai Unigene |
2.2.5 COG功能注释 COG数据库功能注释结果(图 3)形式,共有12 810个Unigene被注释到25个COG功能分类中。其中,翻译、核糖体结构与生物发生的基因最多,有1 525个,占11.9%;其次是一般功能预测基因(1 515个,11.83%),翻译后修饰、蛋白质转运、伴侣基因(1 287个,10.05%),信号传导机制相关基因(1 106个,8.63%),碳水化合物运输和代谢相关基因(1 098个,8.57%)等;注释基因最少的是RNA加工与修饰,仅13个,占0.1%;另有277个Unigene被注释到未知功能基因,762个Unigene与次生代谢生物合成、运输和分解代谢相关,占5.95%。

|
| 图 3 红脚艾Unigene的COG功能分类 Fig. 3 Classification of COG function of Artemisia rubripes Nakai Unigene |
2.3 SSR分布
利用MISA软件筛选得到10 00 bp以上的红脚艾Unigene并进行SSR分析,结果(图 4)鉴定出6种类型、3 529个SSR标记。其中,单碱基重复SSR(p1)最多,共检测到1 684个位点;其次是三碱基重复SSR(p3)共检测到1 017个,双碱基重复SSR(p2) 有539个,复合型重复SSR(c)有232个,四碱基重复SSR(p4)有37个,五碱基重复SSR(p5)和六碱基重复SSR(p6) 较少,分别为5个和11个。

|
| p1:单碱基重复SSR,p2:双碱基重复SSR,p3:三碱基重复SSR,p4:四碱基重复SSR,p5:五碱基重复SSR,p6:六碱基重复SSR,c:复合型重复SSR p1: Mononucleotide repeat SSR, p2: Dinucleotide repeats SSR, p3: Trinucleotide repeats SSR, p4: Quadruple nucleotide repeats SSR, p5: Pentanucleotide repeats SSR, p6: Hexanucleotide repeats SSR, c: Polynucleotide repeats SSR 图 4 红脚艾Unigene的SSR位点分析 Fig. 4 SSR sites analysis of Artemisia rubripes Nakai Unigene |
3 讨论
红脚艾历史上主要分布于岭南地区,惠州市的博罗县、罗浮山及广州市番禺区等地皆有县志及通史记载,但近代以来对其研究与利用很少。随着本草组学技术的发展,许多药用植物基因组得到解析,也使得很多资源得以利用和保护[19]。由于药用植物基因组学研究晚于大众作物,因此转录组学成了本草基因组学研究的重要手段之一。本研究借助高通量测序技术对红脚艾转录组进行了初步探究,共获得7.24 Gb clean data,GC含量为44.67%,Q30碱基百分比达到92.57% 以上;通过Trinity进行de novo组装,共得到173 093个转录本,对组装的转录本去冗余后共获得85 991个Unigene,平均长度为616.87 bp,N50为925 bp,说明红脚艾的组装质量较好,能够满足后续数据分析的要求。
通过数据库比对,注释到47 216个Unigene,与向日葵(H. annuus)、莴苣(L. sativa)、洋蓟(C. cardunculus var. scolymus)等菊科植物匹配基因最多,另有45.09% 的Unigene未被注释,可能由于蒿属植物组学报道较少或组装的序列较短等原因导致[20]。在KEGG代谢通路分析中,核糖体途径、蛋白质加工、氨基酸生物合成途径注释的基因较丰富,可能与氨基酸种类、总氨基酸含量较高有关,而参与类黄酮生物合成的基因有49个,仅占0.29%,这可能是红脚艾侧柏酮含量极低的原因。本研究结果可为红脚艾功能基因挖掘、次级代谢物质合成通路解析提供帮助,也可为中药指纹图谱构建和种质资源鉴定保存及分子辅助良种选育提供基础[21]。
SSR分子标记具有丰度高、共显性和多等位性等优点[22],可利用现有的数据库资源开发SSR引物。本研究鉴定到红脚艾SSR位点3 529个,可为其分子标记开发、遗传多样性分析、种质资源鉴定与优选、分子标记育种等奠定理论基础,为利用分子手段鉴定、区分红脚艾品种及评价其质量提供依据。
4 结论本研究通过高通量测序技术获得了红脚艾的转录组信息特征,经de novo组装共获得85 991个Unigene,有47 216个Unigene在NR、KEGG、COG、KOG、GO和Pfam数据库获得功能注释。KEGG代谢通路中,有16 846个Unigene参与氨基酸、萜类、黄酮类等130条代谢途径,其中有466个Unigene参与氨基酸合成、49个Unigene参与黄酮类合成。同时在红脚艾中还鉴定到3 529个SSR标记,通过获得的红脚艾遗传标记位点分布和Unigene,可进行SSR引物设计,为红脚艾种质资源遗传改良、分子辅助育种奠定基础。
| [1] |
[s1] 国家药典委员会. 中华人民共和国药典: 四部[S]. 北京: 中国医药科技出版社, 2020: 130. National Pharmacopoeia Committee. Pharmacopoeia of the people's Republic of China: Volume Ⅳ[S]. Beijing: China Pharmaceutical Science and Technology Press, 2020: 130. |
| [2] |
简梨娜, 宋学丽, 郭江涛, 刘杰. 艾草的化学成分及临床应用[J]. 化学工程师, 2021, 35(7): 58-62. DOI:10.16247/j.cnki.23-1171/tq.20210758 JIAN L N, SONG X L, GUO J T, LIU J. Chemical composition and clinical application of Artemisia argyi[J]. Chemical Engineer, 2021, 35(7): 58-62. DOI:10.16247/j.cnki.23-1171/tq.20210758 |
| [3] |
张树军, 马耀玲, 王金兰, 李军, 赵明, 白丽明. 艾蒿化学成分研究[J]. 中草药, 2019(8): 9. DOI:10.7501/j.issn.0253-2670.2019.08.020 ZHANG S J, MA Y L, WANG J L, LI J, ZHAO M, BAI L M. Chemical constituents of Artemisia argyi[J]. Chinese Traditional and Herbal Drugs, 2019(8): 9. DOI:10.7501/j.issn.0253-2670.2019.08.020 |
| [4] |
赵秀玲, 党亚丽. 艾叶挥发油化学成分和药理作用研究进展[J]. 天然产物研究与开发, 2019, 31(12): 2182-2188. DOI:10.16333/j.1001-6880.2019.12.023 ZHAO X L, DANG Y L. Advance on chemical constituents and pharmacological effects of Artemisia argyi volatile oil[J]. Nat Prod Res Dev, 2019, 31(12): 2182-2188. DOI:10.16333/j.1001-6880.2019.12.023 |
| [5] |
胡倩, 刘大会, 曹艳. 艾叶黄酮类化合物的研究进展[J]. 食品安全质量检测学报, 2019, 10(12): 16-21. HU Q, LIU D H, CAO Y. Research progress on flavonoids from Artemisia argyi[J]. Journal of Food Safety & Quality, 2019, 10(12): 16-21. |
| [6] |
胡倩, 李静, 刘大会, 曹艳. 艾叶总黄酮提取物体内外抗氧化活性研究[J]. 食品工业科技, 2021, 42(6): 304-309. DOI:10.13386/j.issn1002-0306.2020070227 HU Q, LI J, LIU D H, CAO Y. Antioxidant activity of total flavonoids extracts from folium of Artemisia eargyi in vitro and in vivo[J]. Science and Technology of Food Industry, 2021, 42(6): 304-309. DOI:10.13386/j.issn1002-0306.2020070227 |
| [7] |
梅全喜. 艾叶的研究与应用[M]. 北京: 中国中医药出版社, 2017: 56-103. MEI Q X. Research and application of Artemisia vulgaris[M]. Beijing: China Traditional Chinese Medicine Press, 2017: 56-103. |
| [8] |
邢澍祺, 李杰, 韩惠娟, 周振, 黄建香, 林泽斌, 杨崇仁. 红脚艾营养成分分析与评价[J]. 食品工业科技, 2021, 42(3): 315-319. XING S Q, LI J, HAN H J, ZHOU Z, HUANG J X, LIN Z B, YANG C R. Nutritional components analysis and assessment of Artemisia verlotorum Lamotte[J]. Science and Technology of Food Industry, 2021, 42(3): 315-319. |
| [9] |
HAAS B J, PAPANICOLAOU A, YASSOUR M, GRABHERR M, BLOOD P D, BOWDEN J, COUGER M B, ECCLES D, LI B, LIEBER M, MACMANES M D, OTT M, ORVIS J, POCHET N, STROZZI F, WEEKS N, WESTERMAN R, WILLIAM T, DEWEY C N, HENSCHEL R, LEDUC R D, FRIEDMAN N, REGEV A. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis[J]. Nature Protocols, 2013, 8(8): 1494. DOI:10.1038/nprot.2013.084 |
| [10] |
LI B, DEWEY C N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome[J]. BMC Bioinformatics, 2011, 12(1): 323. DOI:10.1186/1471-2105-12-323 |
| [11] |
ALTSCHUL S F, MADDEN T L, SCHÄFFER A A, ZHANG J, ZHANG Z, MILLER W, LIPMAN D J. Gapped BLAST and PSI BLAST: A new generation of protein database search programs[J]. Nucleic Acids Research Italic, 1997, 25(17): 3389-3402. DOI:10.1093/nar/25.17.3389 |
| [12] |
DENG Y Y, LI J Q, WU S F, ZHU Y, CHEN Y, HE F C. Integrated nr Database in protein annotation system and its localization[J]. Computer Engineering Italic, 2006, 32(5): 71-74. DOI:10.1109/INFOCOM.2006.241 |
| [13] |
APWEILER R, BAIROCH A, WU C H, BARKER W C, BOECKMANN B, FERRO S, GASTEIGER E, HUANG H Z, LOPEZ R, MAGRANE M, MATIN M J, NATALE D A, O'DONOVAN C, REDASCHI N, YEH L S. UniProt: the universal protein knowledgebase[J]. Nucleic Acids Research Italic, 2004, 32: 115-119. |
| [14] |
ASHBURNER M, BALL C A, BLAKE J A, BOTSTEIN D, BUTLER H, CHERRY J M, DAVIS A P, DOLINSKI K, DWIGHT S S, EPPIG J T, HARRIS M A, HILL D P, LAURIE L T, KASARSKIS A, LEWIS S, MATESE J C, RICHARDSON J E, RINGWALD M, RUBIN G M, SHERLOCK. Gene ontology: tool for the unification of biology[J]. Nature Genetics Italic, 2000, 25(1): 25-29. |
| [15] |
TATUSOY R L, GALPERIN M Y, NATALE D A, KOONIN E V. The COG database: a tool for genome scale analysis of protein functions and evolution[J]. Nucleic Acids Research Italic, 2000, 28(1): 33-36. DOI:10.1093/nar/28.1.33 |
| [16] |
KOONIN E V, FEDOROVA N D, JACKSON J D, JACOBS A R, KRYLOY D M, MAKAROVA K S, MAZUMDER R, MEKHEDOW S L, NIKOLSKAYA A N, RAO B S, ROGOZIN L B, SMIRNOV S, SOROKIN A V, SVERDLOV A V, VASUDEVAN S, WOLF Y L, YIN J J, NATALE D A. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes[J]. Genome Biology Italic, 2004, 5(2): 7. DOI:10.1186/gb-2004-5-2-r7 |
| [17] |
KANEHISA M, GOTO S, KAWASHINA S, OKUNO Y. The KEGG resource for deciphering the genome[J]. Nucleic Acids Research Italic, 2004, 32: 277-280. DOI:10.1093/nar/gkh063 |
| [18] |
FINN R D, BATEMAN A, CLEMEMTS J, COGGILL P, EBERHARDT R Y, EDDY S R, HEGER A, HETHERINGTON K, HOLM L, MISTRY J, SONNHAMMER E L L, TATE J, PUNTA M. Pfam: the protein families database[J]. Nucleic Acids Research Italic, 2014, 42: 222-230. DOI:10.1093/nar/gkt1223 |
| [19] |
徐世强, 梅瑜, 曹阳, 黄志娜, 蔡时可, 王继华. 龙脷叶转录组分析及黄酮类生物合成相关基因的挖掘[J]. 广东农业科学, 2020, 47(7): 36-44. DOI:10.16768/j.issn.1004-874X.2020.07.005 XU S Q, MEI Y, CAO Y, HUANG Z N, CAI S K, WANG J H. Analysis of transcriptome and exploration of genes related to flavonoid biosynthesis in Sauropus spatulifolius Beille[J]. Guangdong Agricultural Sciences, 2020, 47(7): 36-44. DOI:10.16768/j.issn.1004-874X.2020.07.005 |
| [20] |
LI D J, DENG Z, QIN B, LIU X H, MEN Z H. De novo assembly and characterization of bark transcriptome using Illumina sequencing and development of EST-SSR markers in rubber tree(Hevea brasiliensis Muell. Arg.)[J]. BMC Genomics, 2012, 13(1): 192. DOI:10.1186/1471-2164-13-192 |
| [21] |
徐世强, 陈永忠, 苏靖朗, 梅瑜, 蔡时可, 陈海燕, 王继华. 黄金艾蒿的非靶向代谢组和转录组学分析[J]. 中药材, 2020, 43(5): 1081-1086. DOI:10.13863/j.issn1001-4454.2020.05.007 XU S Q, CHEN Y Z, SU J L, MEI Y, CAI S K, CHEN H Y, WANG J H. Non-targeted metabolomics and transcriptomics analysis of Artemisia vulgaris 'Variegate'[J]. Journal of Chinese Medicinal Materials, 2020, 43(5): 1081-1086. DOI:10.13863/j.issn1001-4454.2020.05.007 |
| [22] |
周丽霞, 肖勇, 杨耀东. 油棕转录组SSR标记开发研究[J]. 广东农业科学, 2014, 41(14): 136-138, 143. DOI:10.16768/j.issn.1004-874X.2014.14.023 ZHOU L X, XIAO Y, YANG Y D. Development of SSR markers in oil palm(Elaeis guineensis)based on information from transcriptome sequencing[J]. Guangdong Agricultural Sciences, 2014, 41(14): 136-138, 143. DOI:10.16768/j.issn.1004-874X.2014.14.023 |
(责任编辑 崔建勋)